
As filed with the Securities and Exchange Commission on November 22, 2019
Registration No. 333-233281
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
AMENDMENT NO. 2
TO
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
CARDAX, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 2834 | 45-4484428 | ||
(State of incorporation) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification Number) |
2800 Woodlawn Drive, Suite 129
Honolulu, Hawaii 96822
(808) 457-1400
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
David G. Watumull
President and Chief Executive Officer
Cardax, Inc.
2800 Woodlawn Drive, Suite 129
Honolulu, Hawaii 96822
(808) 457-1400
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
Richard M. Morris, Esq. Allegaert Berger & Vogel LLP 111 Broadway, 20th Floor New York, New York 10006 (212) 571-0550
|
Barry I. Grossman, Esq. Sarah E. Williams, Esq. Ellenoff, Grossman & Schole LLP 1345 Avenue of the Americas, New York, NY 10105 (212) 370-1300 |
Approximate date of commencement of proposed sale to the public: From time to time after the effective date of this Registration Statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. [X]
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. [ ]
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. [ ]
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. [ ]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer [ ] | Accelerated filer [ ] | |
| Non-accelerated filer [X] | Smaller reporting company [X] | |
| Emerging growth company [ ] |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act [ ]
CALCULATION OF REGISTRATION FEE
| Title of each class of securities to be registered | Proposed maximum aggregate offering price(1) | Amount of registration fee(2) | ||||||
| Units consisting of shares of Common Stock, par value $0.001 per share, and warrants to purchase shares of Common Stock, par value $0.001 per share | $ | 17,250,000 | (3) | $ | 2,090.70 | |||
| Common stock included as part of the Units | - | - | ||||||
| Warrants to purchase common stock included as part of the Units(4) | - | - | ||||||
| Common stock underlying Warrants | $ | 17,250,000 | $ | 2,090.70 | ||||
| Representative’s Warrants(5) | - | - | ||||||
| Common stock underlying Representative’s Warrants(5) | $ | 970,312.50 | $ | 206.98 | ||||
| Total | $ | 35,470,312.50 | $ | 4,388.38 | ||||
| (1) | Estimated solely for the purpose of calculating the amount of the registration fee pursuant to Rule 457(o) under the Securities Act of 1933, as amended. |
| (2) | Calculated pursuant to Rule 457(o) based on an estimate of the proposed maximum aggregate public offering price. The registration fee has been previously paid. |
| (3) | Equal to $15,000,000 of securities to be offered by us plus the underwriter’s option to purchase up to an additional 15% of the total number of shares offered by us, or up to an additional $2,250,000 of securities, at the public offering price, less underwriting discounts, to cover over-allotments, if any, within 45 days after the date of this prospectus. |
| (4) | No separate registration fee is required pursuant to Rule 457(g) under the Securities Act. |
| (5) | We have agreed to issue upon the closing of this offering, warrants to Maxim Group LLC entitling it to purchase up to 4.5% of the aggregate securities sold in this offering. The exercise price of the warrants is equal to 125% of the public offering price of the common stock offered hereby. The warrants are exercisable commencing six (6) months after the date of effectiveness of this Registration Statement and will terminate five (5) years after the date of effectiveness of this Registration Statement |
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act, or until the registration statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. We may not offer these securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
Subject to Completion
Preliminary Prospectus November 22, 2019
P R O S P E C T U S
_______ Units
Each Unit Consisting of _______ Shares of Common Stock and
_______ Warrants to Purchase _______ Shares of Common Stock
This prospectus relates to our offering of ______ units of Cardax, Inc., a Delaware Corporation (the “Units”). Each unit consists of ______ shares of our common stock and ______ warrants (a “Purchase Warrant”) to purchase ______ shares of our common stock at an exercise price of $______ per share and will expire five years from the date of issuance. The Units will be sold at this fixed price per Unit for the duration of this offering. The public offering price for the Units was determined through negotiations with the underwriters. The Units will not be certificated and the shares of common stock and the Purchase Warrants are immediately separable and will be issued separately in this offering.
Our common stock is quoted for trading on the OTCQB Marketplace (the “OTCQB”) under the symbol “CDXI.” As of November 20, 2019, the last reported sales price for our common stock as quoted on the OTCQB was $0.07 per share. There is no established trading market for the Purchase Warrants. Quotes of stock trading prices on an over-the-counter marketplace may not be indicative of the market price on a national securities exchange. We have applied to have our common stock and Purchase Warrants listed on the Nasdaq Capital Market (the “Nasdaq”) under the symbols “CDXI” and “CDXIW,” respectively. We believe that upon the completion of the offering contemplated by this prospectus, we will meet the standards for listing on the Nasdaq Capital Market. We cannot guarantee that we will be successful in listing our common stock or our Purchase Warrants on Nasdaq, or, if successful, that an active trading market for our common stock or Purchase Warrants will develop or be sustained.
The share and per share information in this prospectus does not reflect the proposed reverse stock split of the issued and outstanding shares of our common stock of ____-for-1 to occur on or prior to the effective date of the offering. The number of shares of common stock will be determined primarily on the basis of the pricing of our Units in this offering. This prospectus will be amended by an amendment to this registration statement to reflect such number and the effect of such reverse stock split, except that we will not reflect the reverse stock split in our Financial Statements and the Notes thereto.
__________________________
An investment in our securities involves a high degree of risk. Before buying any securities you should carefully read the discussion of the material risks of investing in our securities in “Risk Factors” beginning on page 12 of this prospectus.
__________________________
Neither the Securities and Exchange Commission nor any other state securities commission has approved or disapproved of these securities or passed upon the accuracy or adequacy of this prospectus. Any representation to the contrary is a criminal offense.
| Per Unit | Total (Not Including Over-Allotment) | Total (Including Over-Allotment) | ||||||||||
| Public offering price | ||||||||||||
| Underwriting discounts(1) | ||||||||||||
Proceeds to us, before fees and expenses | ||||||||||||
| (1) | We refer you to “Underwriting” beginning on page 65 for additional information regarding total underwriting compensation. |
The underwriters may also purchase up to an additional _________ shares of common stock and/or Purchase Warrants from us at the public offering price, less the underwriting discounts payable by us, to cover over-allotments, if any, within forty-five (45) days from the date of this prospectus (the “Over-Allotment Option”).
The underwriters expect to deliver the shares of common stock and Purchase Warrants to investors on or about , 2019.
__________________________
Sole Book-Running Manager
Maxim Group LLC
The date of this prospectus is_________ , 2019.
TABLE OF CONTENTS
This prospectus is part of a registration statement that we filed with the Securities and Exchange Commission. You should rely only on the information contained in this prospectus or to which we have referred you. Neither we nor the underwriters have authorized anyone to provide any information or to make any representations other than those contained in this prospectus. We take no responsibility for, and can provide no assurance as to the reliability of, any other information that others may give you. This prospectus is an offer to sell only the securities offered hereby, but only under circumstances and in jurisdictions where it is lawful to do so. The information contained in this prospectus is current only as of its date.
Through and including (the 25th day after the date of this prospectus), all dealers effecting transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to a dealer’s obligation to deliver a prospectus when acting as an underwriter and with respect to an unsold allotment or subscription.
For investors outside the U.S.: Neither we nor any of the underwriters have done anything that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the U.S. You are required to inform yourselves about, and to observe any restrictions relating to, this offering and the distribution of this prospectus.
We have not authorized anyone to provide any information or to make any representations other than those contained in this prospectus. We take no responsibility for, and can provide no assurance as to the reliability of, any other information that others may give you. This prospectus is an offer to sell only the shares offered hereby, but only under circumstances and in jurisdictions where it is lawful to do so. The information contained in this prospectus is current only as of its date.
| i |
FORWARD-LOOKING STATEMENTS
There are statements in this prospectus that are not historical facts. These “forward-looking statements” can be identified by use of terminology such as “anticipate,” “believe,” “estimate,” “expect,” “hope,” “intend,” “may,” “plan,” “positioned,” “project,” “propose,” “should,” “strategy,” “will,” or any similar expressions. You should be aware that these forward-looking statements are subject to risks and uncertainties that are beyond our control, including those summarized in this prospectus, such as our ability to develop and commercialize or otherwise monetize our pharmaceutical product candidates as planned, the impact of changes in healthcare regulation, and our ability to raise additional capital to fund our pharmaceutical development activities. For a discussion of these risks, you should read this entire prospectus carefully, especially the risks discussed in the section entitled “Risk Factors.” Although we believe that our assumptions underlying such forward-looking statements are reasonable, we do not guarantee our future performance, and our actual results may differ materially from those contemplated by these forward-looking statements. Our assumptions used for the purposes of the forward-looking statements specified in the following information represent estimates of future events and are subject to uncertainty as to possible changes in economic, legislative, industry, and other circumstances, including the development, acceptance and sales of our products and our ability to raise additional funding sufficient to implement our strategy. As a result, the identification and interpretation of data and other information and their use in developing and selecting assumptions from and among reasonable alternatives require the exercise of judgment. In light of these numerous risks and uncertainties, we cannot provide any assurance that the results and events contemplated by our forward-looking statements contained in this prospectus will in fact transpire. These forward-looking statements are not guarantees of future performance. You are cautioned to not place undue reliance on these forward-looking statements, which speak only as of their dates. We do not undertake any obligation to update or revise any forward-looking statements, except as required by law.
CAUTIONARY NOTE REGARDING INDUSTRY DATA
Unless otherwise indicated, information contained in this prospectus concerning our company, our business, the services we provide and intend to provide, our industry and our general expectations concerning our industry are based on management estimates. Such estimates are derived from publicly available information released by third party sources, as well as data from our internal research, and reflect assumptions made by us based on such data and our knowledge of the industry, which we believe to be reasonable.
| ii |
This summary highlights selected information contained elsewhere in this prospectus and does not contain all the information that you should consider before making your investment decision. Before investing in our common stock and warrants to purchase common stock, you should carefully read this entire prospectus, including the information set forth in the “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” sections of this prospectus and our consolidated financial statements and the accompanying notes included in this prospectus. Except as otherwise indicated herein or as the context otherwise requires, references in this prospectus to “Cardax,” the “Company,” “we,” “us,” and “our” refer to Cardax, Inc. together with its wholly-owned subsidiary, Cardax Pharma, Inc., a Delaware corporation (“Pharma”), and Pharma’s predecessor, Cardax Pharmaceuticals, Inc., a Delaware corporation (“Holdings”), which merged with and into Cardax, Inc. on December 30, 2015 (the “Holdings Merger”). Unless otherwise noted, references in this prospectus to our “product” or “products” includes our pharmaceutical candidates, dietary supplements, and any of our other current or future products, product candidates, and technologies, to the extent applicable.
Overview
We are a development stage biopharmaceutical company focused primarily on the development of pharmaceuticals to safely address one of the major underlying causes of many chronic diseases – inflammation – including cardiovascular disease, metabolic disease, liver disease, arthritis, and aging. We also have a commercial business unit that markets dietary supplements for inflammatory health. We believe we are well positioned for growth through the utilization of astaxanthin and zeaxanthin for chronic pharmaceutical applications by safely reducing chronic inflammation at the cellular and mitochondrial level – without inhibiting normal function. Similar mechanisms also support the use of our dietary supplement for inflammatory health.
We believe that our pharmaceutical product candidates and our dietary supplements have competitive advantages, primarily relating to a unique combination of the following benefits:
| ● | An excellent safety profile that supports chronic use | |
| ● | Broad anti-inflammatory activity and pleiotropic effects with potential application to several chronic diseases as pharmaceuticals and various areas of health as dietary supplements | |
| ● | Oral dosing convenience | |
| ● | Scalable manufacturing | |
| ● | Economical pricing |
Market Overview
There is broad acceptance in the scientific, medical, and financial communities that chronic inflammation is a significant factor in many chronic diseases, particularly cardiovascular disease. The double-blind, randomized, placebo-controlled CANTOS clinical trial (10,061 patients; Novartis, 2017) and REDUCE-IT clinical trial (8,179 patients; Amarin Corporation, 2018), both published in the New England Journal of Medicine, helped to catalyze and support this consensus. Commonly used anti-inflammatory drugs such as aspirin, ibuprofen, naproxen, COX-2 inhibitors, corticosteroids, and various biologics may reduce inflammation, but they have risks of significant side effects that limit their utility in chronic disease.
We believe that a safe anti-inflammatory is the solution. Our lead pharmaceutical candidate CDX-101, a proprietary prodrug of the naturally occurring marine molecule astaxanthin, may provide the needed combination of an excellent safety profile, anti-inflammatory activity, and economic pricing to become widely used for the prevention and treatment of chronic diseases driven by inflammation.
We are pursuing an initial indication of severe hypertriglyceridemia (triglycerides ≥ 500 mg/dL) for CDX-101. Severe hypertriglyceridemia is associated with chronic inflammation and patients with the disorder have increased cardiovascular disease risk and incidence of pancreatitis. We believe the clinical pathway to U.S. Food and Drug Administration (“FDA”) drug approval for severe hypertriglyceridemia, which relies on biomarker endpoints (i.e., measuring triglycerides in blood tests over a period of several months), will be more efficient than other potential indications that require clinical outcomes studies (e.g., evaluating heart attacks, strokes, and deaths over a period of several years), and is thus better suited as our initial indication for CDX-101.
An estimated 3.4 million Americans have severe hypertriglyceridemia according to peer-reviewed research published in the American Journal of Cardiology in 2011. Statins, fibrates, and prescription fish oils are all used to manage hypertriglyceridemia. 21% (42 million) of U.S. adults have mixed dyslipidemia (high levels of low-density lipoprotein “LDL” cholesterol with low levels of high-density lipoprotein “HDL” cholesterol and/or high levels of triglycerides), with nearly 6% (11.6 million people) having all three lipid abnormalities. Lovaza, Vascepa, and other prescription fish oils approved for severe hypertriglyceridemia are also used off-label in mixed dyslipidemia patients to reduce moderately elevated triglycerides and aggregate sales of these products for on and off-label use are estimated to be approaching $2 billion annually.
We believe CDX-101 will have several competitive advantages compared to prescription fish oils: (i) ease of administration: oral dosing of large fish oil capsules is problematic, whereas we expect CDX-101 tablets should be far smaller; (ii) scalability: prescription fish oil manufacturing is limited by the declining global fish supply, whereas we believe the synthetic production of CDX-101 is scalable; and (iii) safety: prescription fish oils have certain safety risks, whereas we believe that astaxanthin, the active moiety of CDX-101, has an excellent safety profile.
The REDUCE-IT clinical trial demonstrated that administration of Vascepa resulted in a significant reduction of major adverse cardiovascular events (“MACE”) in patients with mixed dyslipidemia on standard of care, specifically statins, and we believe is the primary basis of Amarin’s request to the FDA to expand Vascepa’s label. The reduction of triglycerides in the REDUCE-IT clinical trial was modest however, and the study’s authors concluded that Vascepa’s ability to reduce other markers of cardiovascular disease, including inflammation and oxidized LDL (as demonstrated in the MARINE and ANCHOR clinical trials), provided the pleiotropic effects that led to reduction of MACE in REDUCE-IT. In human proof-of-concept “pilot” studies conducted by third parties and animal models conducted by third parties and us, astaxanthin, the active moiety of CDX-101, has demonstrated similar pleiotropic effects, which are derived from its broad anti-inflammatory activity, but without the limitations of Vascepa or other prescription fish oils. As a result, we believe this market also presents a major opportunity as a potential second indication for CDX-101.
| 1 |
Beyond cardiovascular disease, we believe CDX-101 could be developed to address other chronic diseases driven by inflammation, including metabolic disease, liver disease, arthritis, and aging, each with potential annual sales exceeding a billion dollars.
We are also developing CDX-301, our zeaxanthin pharmaceutical candidate, for macular degeneration. Our target initial indication for CDX-301 is Stargardt disease, a juvenile form of macular degeneration and potential orphan drug indication. Zeaxanthin has a mechanism of action and excellent safety profile similar to astaxanthin, however, it accumulates in the human eye through uptake by a unique retinal receptor, providing protection against blue light, oxidative damage, and related inflammation that occurs in macular degeneration. Pre-clinical and clinical studies with zeaxanthin have demonstrated proof-of-concept for the treatment of macular disorders. Based on multiple academic and NIH sources, we believe there are no more than 42,000 persons in the United States with Stargardt disease, and therefore we believe a treatment for Stargardt disease may qualify for orphan drug designation. (By statute, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition that affects less than 200,000 persons in the United States.) If CDX-301 receives FDA orphan drug designation for Stargardt disease and obtains FDA drug approval, we expect CDX-301 may benefit from certain advantages as an orphan drug, including orphan drug exclusivity, which means the FDA may not approve any other application, including a full new drug application (“NDA”), to market the same drug for the same indication for a period of seven years, except in limited circumstances. We also believe that age related macular degeneration, a larger market estimated to afflict more than three million people in the U.S. alone, presents a major opportunity as a potential second indication for CDX-301. We do not expect to use the proceeds of this offering to pursue the development of CDX-301.
Astaxanthin
Astaxanthin Safety
Astaxanthin is a naturally occurring marine carotenoid found in salmon, microalgae, krill, lobster, and crab. Carotenoids are natural pigments that impart coloration and support animal health and vitality, especially in harsh marine environments. Astaxanthin is responsible for the characteristic red or pink color of salmon and shellfish. Salmon without astaxanthin are smaller, more susceptible to infection, have reproductive problems, and are not strong enough to swim upstream.
Astaxanthin is Generally Recognized as Safe (“GRAS”) as a food substance according to FDA regulations and has undergone extensive toxicity testing by third parties and us with no clinically meaningful issues even at the extremely high doses summarized in the table below:
| Type of Study | Maximum Dosing | |
| Acute Toxicity | >8,000 mg/kg (mouse, rat), 2,000 mg/kg (non-human primates) | |
| Sub-Chronic Toxicity | 1,240 mg/kg (rat), 160 mg/kg (dog) | |
| 1 Year Chronic Toxicity/Carcinogenicity | 1,000 mg/kg (rat), 1,400 mg/kg (mouse), 200 mg/kg (dog) | |
| 2 Year Carcinogenicity | 1,000 mg/kg (rat) | |
| Genotoxicity/Mutagenicity | 2,000 mg/kg (mouse) | |
| Teratogenicity | 1,000 mg/kg (rat), 400 mg/kg (rabbit) |
Commonly used anti-inflammatory drugs such as aspirin, ibuprofen, naproxen, COX-2 inhibitors, corticosteroids, and various biologics have risks of side effects including gastrointestinal bleeding, heart attacks, strokes, and severe infections. Prescription fish oil drugs, while safer than common anti-inflammatory drugs, also have risks of certain side effects. Lovaza and other DHA, EPA combination fish oil drugs, have risks of side effects including back pain, eructation, dysgeusia, and increases in LDL cholesterol. Vascepa has risks of side effects including arthralgia, atrial fibrillation, and increased bleeding. Fenofibrates have risks of side effects including stomach pain, nausea, and back pain.
In contrast, astaxanthin has no known side effects of clinical significance. We believe astaxanthin’s excellent safety profile will be a key competitive advantage compared to other drugs targeting inflammation and lipids.
Astaxanthin Mechanism of Action
The mechanism of action of astaxanthin, the active moiety in CDX-101, is quite different than most drugs, and we believe is responsible for its excellent safety profile. Most drugs target single receptors or enzymes in complex pathways, which can lead to side effects with chronic use. Astaxanthin is distributed systemically, including to the liver and heart, where it localizes in cellular and mitochondrial membranes and reduces the oxidative stress that causes chronic inflammation, without affecting the normal function of inflammatory/metabolic signaling pathways. And unlike other antioxidants such as beta-carotene, Vitamin C, and Vitamin E, astaxanthin spans and stabilizes cellular and mitochondrial membranes (biological lipid bilayers) to function as an aqueous and lipid phase antioxidant without membrane disruption, as proven by X-ray diffraction studies:
| 2 |
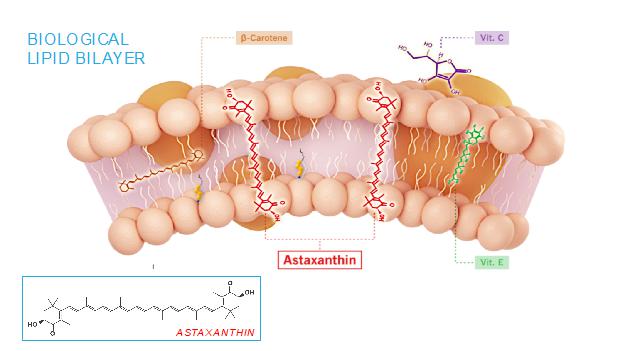
As a result, astaxanthin demonstrates positive and quantifiable pleiotropic effects on many inflammatory cytokines and drug targets.
In human proof-of-concept “pilot” studies conducted by third parties, astaxanthin statistically significantly decreased inflammation and oxidative stress:
| ● | TNF-α decreased (-30%, p=0.0022) | |
| ● | C-Reactive Protein (“CRP”) decreased (-20%, p<0.05; two studies) | |
| ● | Oxidative stress decreased (MDA, IsoP, SOD, TAC increased) |
In animal studies conducted by third parties, astaxanthin statistically significantly decreased inflammation and oxidative stress:
| ● | Inflammatory markers decreased in various model systems: |
| ○ | TNF-α, IL-1β, IL-6, CRP, NF-kB, PGE-2, iNOS, MCP-1, MPO, ERK, JNK, COX-2 | ||
| ○ | TNF-α decreased equivalent to an equal dose of prednisolone |
| ● | Oxidative stress decreased in mitochondria |
Astaxanthin Research Results
There are more than 2,000 published peer reviewed papers related to astaxanthin, including more than 50 peer reviewed papers published by Cardax and its collaborators (referred to herein as “us”) and more than 50 “pilot” human clinical trials with astaxanthin supplements, more than 20 of which were randomized, double-blind, placebo-controlled human proof-of-concept studies. As discussed in greater detail under “Business – Astaxanthin Research Results” on page 37 of this prospectus, highlights of astaxanthin’s pleiotropic effects, which were demonstrated in studies utilizing astaxanthin from natural and synthetic sources, include:
| ● | Astaxanthin and Cardiovascular Disease: In human proof-of-concept “pilot” studies conducted by third parties, astaxanthin statistically significantly decreased inflammation, triglycerides, LDL cholesterol, and blood pressure. In animal studies conducted by third parties and us, astaxanthin demonstrated statistically significant improvements in models of cardiovascular disease. | |
| ● | Astaxanthin and Metabolic Disease: In human proof-of-concept “pilot” studies conducted by third parties, astaxanthin statistically significantly increased adiponectin and decreased TNF-α and oxidative stress. In animal studies conducted by third parties, astaxanthin demonstrated statistically significant improvements in models of metabolic disease. | |
| ● | Astaxanthin and Liver Disease: In human proof-of-concept “pilot” studies conducted by third parties, astaxanthin statistically significantly decreased fat accumulation in biopsy-diagnosed nonalcoholic steatohepatitis (“NASH”) patients, decreased TNF-α, improved lipid profile parameters, and decreased oxidative stress. In animal studies conducted by third parties and us, astaxanthin statistically significantly decreased elevated liver enzymes, lipids, insulin resistance, steatosis, and fibrosis. | |
| ● | Astaxanthin and Arthritis: In human proof-of-concept “pilot” non-arthritis studies conducted by third parties, astaxanthin statistically significantly decreased markers of inflammation of relevance to arthritis, including TNF-α and CRP. In animal studies conducted by third parties, astaxanthin decreased inflammation, oxidative stress, and joint degeneration. | |
| ● | Astaxanthin and Aging: In human studies conducted by third parties, activation of the FOXO3 gene has been linked to decreased inflammation and aging. In animal studies conducted by third parties and us, astaxanthin activated the FOXO3 gene and extended lifespan. |
| 3 |
Our Products and Business Strategy
Our product platform consists of our development stage pharmaceutical candidates and our commercially available dietary supplement:
| ● | CDX-101, our lead pharmaceutical candidate, is in pre-clinical development for cardiovascular inflammation and dyslipidemia, with a target initial indication of severe hypertriglyceridemia. | |
| ● | CDX-301 is in pre-clinical development for macular degeneration, with a target initial indication of Stargardt disease. | |
| ● | ZanthoSyn® is a physician recommended astaxanthin dietary supplement for inflammatory health. |
Lead Pharmaceutical Candidate: CDX-101
Our lead pharmaceutical candidate, CDX-101, is a proprietary astaxanthin prodrug that cleaves following oral administration and delivers astaxanthin to the bloodstream. CDX-101 is being developed initially for cardiovascular inflammation and mixed dyslipidemia, with a target initial indication of severe hypertriglyceridemia.
We believe that the results from two major cardiovascular clinical trials—the 10,061 patient CANTOS study by Novartis in 2017 and the 8,179 patient REDUCE-IT study by Amarin in 2018—clearly demonstrated the clinical significance of reducing chronic inflammation, validating the cardiovascular inflammation hypothesis we have supported for more than a decade. We believe that astaxanthin’s unique mechanism of action—reduction of oxidative stress driven inflammation at the cellular and mitochondrial level without inhibiting normal function—results in an impact on key inflammatory drug targets and pathways, and importantly, an excellent safety profile that supports chronic administration. In addition to the safety advantages described in this prospectus, we believe that production of CDX-101, unlike Vascepa and other prescription fish oil drugs, will be highly scalable to address these large mass markets for chronic diseases driven by inflammation.
Clinical and non-clinical studies with astaxanthin have provided proof-of-concept for the treatment of cardiovascular risk factors including inflammation and triglycerides as described in this prospectus. In addition, interim results from our Cardiovascular Health Astaxanthin Supplement Evaluation (“CHASE”) clinical trial demonstrate beneficial changes in markers of cardiovascular health, including CRP, LDL cholesterol, total cholesterol, triglycerides, oxidized LDL, and blood pressure, and also underscore astaxanthin’s safety profile with no adverse safety signals observed. We believe these findings provide further mechanistic support for our pharmaceutical development program. We refer you to “CHASE Clinical Trial” on page 5 of this prospectus for additional information regarding the CHASE clinical trial.
We believe that an initial indication of severe hypertriglyceridemia provides an efficient clinical pathway to drug approval for CDX-101 and will be similar to the pathway as reported by Amarin for the development of Vascepa, its prescription fish oil. CDX-101 is currently in pre-clinical development, including the planning of Investigational New Drug (“IND”) enabling studies. We plan to use proceeds from this offering to complete IND enabling studies and to engage third party contract development and manufacturing organizations (CDMOs) to manufacture drug substance and drug product for such studies, with the goal of filing an IND approximately one year from the closing of this offering.
We have retained Paresh N. Soni, M.D., Ph.D., the former Senior Vice President and Head of Development at Amarin, to guide our clinical and regulatory strategy, interact with the FDA, and advise us on a full range of development issues. While at Amarin, Dr. Soni led the design of Amarin’s clinical trials, development strategy, and interaction with the FDA, including for Vascepa, which was approved for treatment of severe hypertriglyceridemia in 2012. Dr. Soni played a key role in the design and conduct of the MARINE, ANCHOR, and REDUCE-IT clinical trials with Vascepa. Dr. Soni is also a member of our Scientific Advisory Board.
In addition to Dr. Soni, our Scientific Advisory Board includes Deepak L. Bhatt, M.D., M.P.H. and R. Preston Mason, Ph.D.
Deepak L. Bhatt, M.D., M.P.H., is the Chairman of our Scientific Advisory Board. Dr. Bhatt is also the Chair of the REDUCE-IT clinical trial with Vascepa, Executive Director of Interventional Cardiovascular Programs at Harvard Medical School affiliated Brigham and Women’s Hospital, and Professor at Harvard Medical School. He is also the Editor of the peer-reviewed Journal of Invasive Cardiology and Editor-in-Chief of the Harvard Heart Letter for patients.
R. Preston Mason, Ph.D. is on the faculty of the Department of Medicine, Division of Cardiology at Harvard Medical School affiliated Brigham and Women’s Hospital. He has published more than 250 peer reviewed papers, including papers published in collaboration with Cardax, and is a recognized expert on the mechanism of action of astaxanthin and fish oils, particularly Vascepa.
CDX-101 vs. ZanthoSyn®
CDX-101 is a synthetic astaxanthin prodrug (new chemical entity) for pharmaceutical applications and ZanthoSyn® is a formulation of synthetic nature-identical astaxanthin for dietary supplement applications. While both deliver astaxanthin to the bloodstream, we believe the unique molecular structure of CDX-101 and its pharmaceutical pathway will provide substantial differentiation. In particular, we believe that:
| ● | CDX-101 will be approved by the FDA as a drug for one or more disease indications, whereas ZanthoSyn® is marketed as a dietary supplement for health applications; | |
| ● | CDX-101 will be prescribed by doctors and covered by health insurance, whereas ZanthoSyn® is sold through retail and e-commerce channels; | |
| ● | CDX-101 will be administered at a higher dose and in different oral dosage form; and | |
| ● | CDX-101 will have superior intellectual property protection. |
| 4 |
Pharmaceutical Candidate: CDX-301
Our zeaxanthin pharmaceutical candidate, CDX-301, has a mechanism of action and excellent safety profile similar to astaxanthin, however, it is being developed for macular degeneration because zeaxanthin accumulates in the human eye through uptake by a unique retinal receptor, providing protection against blue light, oxidative damage, and related inflammation that occurs in macular degeneration. Pre-clinical and clinical studies with zeaxanthin have demonstrated proof-of-concept for the treatment of macular disorders. We believe that an initial indication of Stargardt disease, a juvenile form of macular degeneration, provides an efficient clinical pathway to drug approval for CDX-301. On November 30, 2018, we submitted a request for orphan drug designation to the FDA for zeaxanthin as a treatment of Stargardt disease, and we are currently in communications with the FDA regarding this matter. Additional financing beyond that contemplated in this offering will be needed to fund IND enabling studies and clinical development of CDX-301.
Dietary Supplement: ZanthoSyn®
ZanthoSyn® is our commercially available physician recommended astaxanthin dietary supplement. Astaxanthin is a naturally occurring molecule with safe anti-inflammatory activity that supports cardiovascular health, metabolic health, liver health, joint health, and longevity. The form of astaxanthin utilized in ZanthoSyn® has demonstrated an excellent safety profile in peer-reviewed published studies and is GRAS according to FDA regulations.
We sell ZanthoSyn® primarily through wholesale and e-commerce channels. We launched our e-commerce channel in 2016 and began selling to General Nutrition Corporation (“GNC”) stores in 2017. ZanthoSyn® is currently available at GNC corporate stores nationwide in the United States.
ZanthoSyn® is the top selling product at GNC stores in Hawaii and the top selling product in the anti-oxidant category at GNC stores nationwide.
We market ZanthoSyn® primarily through a multi-pronged approach:
| ● | Physician outreach and education, where ZanthoSyn® is positioned as the first safe, physician friendly, anti-inflammatory dietary supplement for health and longevity, with retail locations and e-commerce serving as convenient and credible distribution channels for physicians recommending ZanthoSyn® | |
| ● | Retail store outreach, education, and in-store sales support, building on the ability to utilize ZanthoSyn® as a foundation of health and wellness regimens | |
| ● | E-commerce platforms |
We believe ZanthoSyn® is physician friendly for several reasons:
| ● | ZanthoSyn® delivers the safety, purity, manufacturing rigor, bioavailability, and scientific support that provides physicians comfort in the quality and utility of the product, which is often not present in other dietary supplements. | |
| ● | ZanthoSyn® is well-accepted at medical conferences where crowds of physicians and other healthcare professionals stand in line to receive ZanthoSyn® samples and product information after attending educational seminars. |
Our sales and marketing program was initially launched in Hawaii, where we believe that robust physician outreach and education coupled with GNC retail store outreach, education, and in-store sales support increased consumer awareness and catalyzed strong sales growth. We also launched this program in major markets on the West Coast and East Coast in the U.S. beginning in 2017. To support these efforts, we have hired additional sales and marketing personnel. We are currently evaluating our strategy related to further expansion.
We sell ZanthoSyn® to GNC under a purchasing agreement. The exclusivity provision under such agreement related to distribution of ZanthoSyn® by GNC in the “brick and mortar” retail channel in the United States expired on October 16, 2019. GNC remains our only distributor of ZanthoSyn® in such channel, but we may expand retail distribution to mass market retailers, other specialty nutrition stores, pharmacies, and other retailers. We also plan to increase our sales and marketing efforts through e-commerce.
CHASE Clinical Trial
In September 2018, we initiated a human clinical trial entitled, Cardiovascular Health Astaxanthin Supplement Evaluation (“CHASE”), targeting cardiovascular inflammatory health. The randomized, double-blind, placebo-controlled clinical trial is evaluating the effect of low-dose and high-dose ZanthoSyn® on cardiovascular health as measured by CRP levels over 12 weeks in up to 120 subjects with documented cardiovascular risk factors. The study also includes an optional open label extension through 48 weeks.
Interim results from an initial cohort of subjects were announced on September 23, 2019. The interim results were based on data from 40 subjects administered high-dose ZanthoSyn® (96 mg/day astaxanthin – 48 mg twice a day), low-dose ZanthoSyn® (24 mg/day astaxanthin – 12 mg twice a day), or placebo.
Highlights from the interim review shown below are median percentage changes from baseline to week 12 unless otherwise stated. While the interim review was not powered for statistical significance, p-values less than 0.05 compared to placebo are provided. The p-values reported below (*p<0.05, **p<0.01) are nominal p-values from non-parametric comparisons of the median between each group and placebo and no adjustments for multiple comparisons were made.
| Interim Results | High Dose | Low Dose | Placebo | |||||||||
| CRP | -28% | -32% | -5% | |||||||||
| LDL-C | -12% | ** | -7% | +5% | ||||||||
| Total cholesterol | -8% | * | -5% | +4% | ||||||||
| Triglycerides | -16% | -13% | +6% | |||||||||
| Oxidized LDL | -10% | * | +3% | +4% | ||||||||
| Blood pressure | -5% | * | -4% | * | +6% | |||||||
| Median astaxanthin blood levels at 12 weeks | 2,184 ng/mL | 790 ng/mL | <10 ng/mL | |||||||||
We believe these findings provide:
| ● | Further mechanistic support for our astaxanthin pharmaceutical development program | |
| ● | Basis for additional patent filings | |
| ● | Support for the cardiovascular health benefits of ZanthoSyn® |
The interim results also underscore astaxanthin’s safety profile with no adverse safety signals observed. The CHASE Data Safety Review Board, which is comprised of a majority of independent clinical trial professionals, recommended that the clinical trial continue enrollment.
The FDA does not require human clinical trials for dietary supplements, but we believe that positive results from the CHASE trial may help promote scientific and consumer awareness of astaxanthin’s health and longevity applications and serve as further mechanistic support for our pharmaceutical development program.
We refer to you the “Risk Factors” section of this prospectus for a summary of certain risks related to clinical trial results.
| 5 |
REPZ Clinical Study
We are also exploring the effect of ZanthoSyn® on recovery, endurance, and performance in a clinical study (the Recovery, Endurance, and Performance with ZanthoSyn® or “REPZ” study) with 40 subjects by measuring sprint times and heart rates in connection with high intensity interval training on stationary air bikes. The results of the REPZ study, if successful, may be used to support ZanthoSyn® marketing efforts for sports and fitness applications.
The REPZ study was recently completed, and final data analysis is underway.
Benefits of Synthetic Astaxanthin vs. Natural Astaxanthin
Dietary supplements containing astaxanthin typically derive astaxanthin from microalgae, krill, or other natural sources, whereas ZanthoSyn® astaxanthin is made through total synthesis. While multiple studies demonstrate that astaxanthin from either natural or synthetic sources is efficacious and both are Generally Recognized as Safe according to FDA regulations, we believe synthetic astaxanthin offers significant advantages compared to astaxanthin from microalgae, krill, or other natural sources:
| ● | Synthetic astaxanthin can be formulated for superior bioavailability. In a human crossover study comparing ZanthoSyn® to a leading microalgal astaxanthin dietary supplement, the astaxanthin blood levels following administration of ZanthoSyn® were nearly three times higher than the microalgal astaxanthin product at the same dose: |
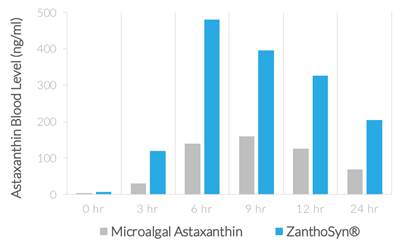
| ● | AUC (area under curve, astaxanthin blood levels) = 2.85-fold greater (p=0.013) | |
| ● | Cmax (maximum concentration, astaxanthin blood levels) = 3.0-fold greater (p=0.013) | |
| ● | Coefficient of variation (variation between subjects of astaxanthin blood levels) |
| o | ZanthoSyn® = 27% | |
| o | Microalgal astaxanthin = 62% |
| ● | Tmax (time of maximum concentration) = 6 hours | |
| ● | No adverse events observed |
The superior bioavailability described in this prospectus means that three times more astaxanthin from ZanthoSyn® is absorbed into the body from each dose, which provides a superior value proposition compared to other astaxanthin dietary supplements.
| ● | Synthetic astaxanthin has been extensively tested in a wide range of toxicity studies, including acute, sub-acute, sub-chronic, and chronic toxicity studies, carcinogenicity studies, genotoxicity/mutagenicity studies, and developmental and reproductive toxicity studies; whereas to our knowledge microalgal or other sources of astaxanthin have not undergone the same amount of safety testing in such toxicity studies. | |
| ● | Synthetic astaxanthin is manufactured with superior purity and precision, whereas astaxanthin extracted from microalgae and krill oil is obtained in a complex mixture, which may include many unknown marine byproducts. | |
| ● | Synthetic manufacture of astaxanthin is scalable, whereas we believe the ability to readily scale the production and extraction of astaxanthin from microalgae or other sources will be limited as demand for astaxanthin grows. | |
| ● | Synthetic manufacture of astaxanthin emits fewer greenhouse gases and consumes less energy, raw material, and land than traditional microalgal astaxanthin production. |
| 6 |
Intellectual Property
We have obtained and are continuing to seek patent protection for compositions of matter, pharmaceutical compositions, and pharmaceutical uses, in certain disease areas, of our various carotenoid analogs and derivatives. Such carotenoids include astaxanthin, zeaxanthin, lutein, and/or lycophyll, and esters and other analogs and derivatives of these compounds. More specifically, we seek to protect: (i) the composition of matter of novel carotenoid analogs and derivatives, (ii) pharmaceutical compositions comprising synthetic or natural preparations of novel or natural occurring carotenoid analogs and derivatives, and (iii) the pharmaceutical use of synthetic preparations of novel or naturally occurring carotenoid analogs and derivatives in specific disease areas, including, but not limited to, the treatment of inflammation and related tissue damage, liver disease, and reperfusion injury, as well as the pharmaceutical use of synthetic or natural preparations of novel or natural occurring carotenoid analogs and derivatives for the reduction of platelet aggregation. We intend to enforce and defend our intellectual property rights consistent with our strategic business objectives.
We have 29 issued patents and two pending patents related to the composition of matter, pharmaceutical compositions, and pharmaceutical uses of our drugs candidates as well as many other related molecules that will expire between 2023 and 2028, subject to patent term extensions. We also have filed additional patents to extend patent coverage in the U.S. and worldwide to 2039-2040, with such applications including coverage related to certain cardiovascular uses on the basis of the CHASE clinical trial results as well as coverage related to the composition of matter of CDX-101.
The Company’s patents are summarized in the table below.
| United States | Foreign | Expiration | ||||||||||
| Issued Patents | 14 | 15 | 2023-2028 | |||||||||
| Pending Patents | 0 | 1 | 2023-2028 | |||||||||
| Pending Patents | 2 | 1 | 2039-2040 | |||||||||
Reverse Stock Split
On October 2, 2019, our stockholders authorized a reverse stock split of our common stock (the “Reverse Stock Split”) within the range that is considered appropriate and necessary for our common stock to have a targeted trading price per share that meets the listing requirements of the Nasdaq Capital Market, at a minimum, and such other price determined appropriate by our Board of Directors (the “Board”), and authorized the Board, in its sole discretion, to determine the final ratio of shares for the Reverse Stock Split on the effective date and to file a certificate of amendment to our amended and restated certificate of incorporation in connection with the Reverse Stock Split.
On ______, the Board established a ratio for the Reverse Stock Split of the issued and outstanding shares of our common stock of ____-for-1 and the Reverse Stock Split was effective at 12:01 a.m. on ______. Trading of our common stock on a post-Reverse Stock Split basis began at market open on ______. No fractional shares were issued in the Reverse Stock Split and any remaining share fractions were rounded up to the next whole share.
In connection with the Reverse Stock Split, the issued and outstanding shares of our common stock were combined by the ______-for-1 ratio. Also, all shares of our common stock subject to outstanding equity awards and the exercise price of any such award (if applicable) and the number of shares remaining available for issuance under the Cardax, Inc. 2014 Equity Compensation Plan as amended, and all shares underlying outstanding warrants, convertible notes, and other derivative securities of the Company, including exercise prices and conversion prices (if applicable) were proportionately adjusted for the Reverse Stock Split.
Corporate Information
We are a development stage biopharmaceutical company engaged in the development and commercialization of pharmaceuticals and dietary supplements. We are a smaller reporting company as defined by applicable federal securities regulations. Our common stock is traded on the OTCQB under the trading symbol “CDXI”. On the effective date of this prospectus, we expect that trading on Nasdaq will be under the same symbol. We also intend to seek a listing for the Purchase Warrants on Nasdaq under the symbol “CDXIW.”
Our executive offices are located at 2800 Woodlawn Drive, Suite 129, Honolulu, Hawaii 96822. Our telephone number is (808) 457-1400. Our website is located at www.cardaxpharma.com. The information on our website or that can be accessed through our website does not constitute a part of this prospectus.
Summary of Risk Factors
Investing in our common stock and warrants to purchase common stock involves substantial risk. You should carefully consider all of the information in this prospectus before investing in our common stock and warrants to purchase common stock, including the risks related to this offering and our common stock, our business and industry, our intellectual property, our financial results, and our need for financing, each as described in the section entitled “Risk Factors” and elsewhere in this prospectus.
| 7 |
Our business is subject to numerous risks, as more fully described in the section entitled “Risk Factors” immediately following this prospectus summary. You should read these risks before you invest in our common stock. In particular, our risks include, but are not limited to, the following:
| ● | We have a history of operating losses, have received a going concern opinion from our auditors, and may not have sufficient funds to complete the development and commercialization of our pharmaceutical candidates. | |
| ● | Our management will have broad discretion as to the use of proceeds from this offering, and we may not use the proceeds effectively. | |
| ● | An active, liquid, and orderly market for our common stock or Purchase Warrants may not develop. | |
| ● | The Purchase Warrants may not have any value. | |
| ● | A number of different factors could prevent us from developing or commercializing our products on a timely basis, or at all. | |
| ● | We operate in highly competitive industries, and our failure to compete effectively could adversely affect our market share, financial condition and growth prospects. If competitors are better able to develop and market products that are more effective, or gain greater acceptance in the marketplace than our products, our commercial opportunities may be reduced or eliminated. | |
| ● | The pharmaceutical and dietary supplement industries are subject to extensive and complex healthcare regulation. Any determination that we have violated federal or state laws applicable to us that regulate healthcare would have a material adverse effect on our business, prospects, and financial condition. | |
| ● | If we fail to comply with FDA regulations our business could suffer. | |
| ● | Orphan drug designation for our products may not confer marketing exclusivity or other expected benefits. | |
| ● | We rely on third parties to supply and manufacture our products. If these third parties do not perform as expected or if our agreements with them are terminated, our business, prospects, financial condition, and results of operations would be materially adversely affected. | |
| ● | Our ability to market our products may be impaired by the intellectual property rights of third parties. | |
| ● | We may be involved in lawsuits or proceedings to protect or enforce our intellectual property rights or to defend against infringement claims, which could be expensive and time consuming. | |
| ● | Our ability to grow and compete in the future will be adversely affected if adequate capital is not available to us or not available on terms favorable to us. | |
| ● | Provisions in our corporate charter documents and under Delaware law could make an acquisition of us more difficult and may prevent attempts by our stockholders to replace or remove our current management. |
| 8 |
The Offering
| Securities offered by us: | ________ units, with each unit consisting of ______ shares of our common stock and ______ Purchase Warrants to purchase ______ shares of our common stock at an exercise price of $______ per share. The Units will not be certificated and the shares of common stock and the Purchase Warrants are immediately separable and will be issued separately in this offering. | |
| Common stock offered by us: | ______ shares. | |
| Purchase Warrants offered by us: | ______ Purchase Warrants, each providing the right to purchase ______ shares of our common stock. Each Purchase Warrant will have an exercise price per share of ______, will be exercisable on the original issuance date, and will expire on the fifth anniversary of the original issuance date. Each holder of Purchase Warrants will be prohibited from exercising its Purchase Warrant for shares of our common stock if, as a result of such exercise, the holder, together with its affiliates, would own more than 4.99% of the total number of shares of our common stock then issued and outstanding. However, any holder may increase such percentage to any other percentage not in excess of 9.99%, provided that any increase in such percentage shall not be effective until 61 days after such notice to us. This prospectus also relates to the offering of the shares of common stock issuable upon exercise of the Purchase Warrants. | |
| Common stock to be outstanding after this offering(1): | ______ shares. | |
| Option to purchase additional shares of our common stock and/or Purchase Warrants: | The underwriters have an option within 45 days of the date of this prospectus to purchase up to ______ additional shares of our common stock and/or Purchase Warrants at the public offering price, less the underwriting discount. | |
| The Nasdaq symbol for our common stock: | Our common stock is currently traded on the OTCQB. In connection with this offering, we expect to have our shares of our common stock listed for trading on the Nasdaq Capital Market under the symbol “CDXI”. | |
| Proposed Nasdaq listing for Purchase Warrants: | There is no established public trading market for the Purchase Warrants. We intend to seek a listing for the Purchase Warrants on Nasdaq under the symbol “CDXIW,” however we cannot assure you that we will be successful listing the Purchase Warrants on Nasdaq or, if successful, that an active trading market for the Purchase Warrants will develop or be sustained. | |
| Use of proceeds: | We estimate the net proceeds to us from the sale of ______ Units at an assumed combined public offering price of $______ per Unit will be approximately $_________ million after deducting underwriting discounts and estimated offering fees and expenses payable by us. If the underwriters exercise their option to purchase additional shares of our common stock and/or Purchase Warrants in full, we estimate that our net proceeds will be approximately $_________ million after deducting underwriting discounts and estimated offering fees and expenses payable by us. | |
We intend to use the net proceeds from the sale of the Units to fund our research, development, and clinical programs, including the funding of our budgeted expenditures to develop our CDX-101 pharmaceutical candidate through IND and to complete our CHASE clinical trial targeting cardiovascular inflammatory health with our ZanthoSyn® astaxanthin dietary supplement, as well as for other general corporate purposes, including working capital and repayment of certain indebtedness. See “Use of Proceeds” on page 25 of this prospectus. | ||
| Lock-ups: | We, our officers and directors, and certain holders of our capital stock will enter into lock-ups restricting the transfer of shares of or relating to our capital stock for six (6) months after the date of this prospectus. |
| 9 |
| Reverse Stock Split: | On October 2, 2019, our stockholders authorized a Reverse Stock Split within the range that is considered appropriate and necessary for our common stock to have a targeted trading price per share that meets the listing requirements of the Nasdaq Capital Market, at a minimum, and such other price determined appropriate by our Board, and authorized the Board, in its sole discretion, to determine the final ratio of shares for the Reverse Stock Split on the effective date and to file a certificate of amendment to our amended and restated certificate of incorporation in connection with the Reverse Stock Split. | |
On ______, the Board established a ratio for the Reverse Stock Split of the issued and outstanding shares of our common stock of ____-for-1 and the Reverse Stock Split was effective at 12:01 a.m. on ______. Trading of our common stock on a post-Reverse Stock Split basis began at market open on ______. No fractional shares were issued in the Reverse Stock Split and any remaining share fractions were rounded up to the next whole share. | ||
| In connection with the Reverse Stock Split, the Company’s issued and outstanding shares were combined by the ______-for-1 ratio. Also, all shares of our common stock subject to outstanding equity awards and the exercise price of any such award (if applicable) and the number of shares remaining available for issuance under the Cardax, Inc. 2014 Equity Compensation Plan, as amended, and all shares underlying outstanding preferred stock and other derivative securities of the Company, including exercise prices and conversion rates (if applicable) were proportionately adjusted for the Reverse Stock Split. | ||
| Risk factors: | You should read the “Risk Factors” section beginning on page 12 and other information included in this prospectus for a discussion of factors to consider carefully before deciding to invest in our securities. |
| (1) | The number of shares of our common stock outstanding immediately after this offering is based on ______ shares of our common stock outstanding as of September 30, 2019, after giving effect to the Reverse Stock Split, and excludes: |
| ● | ______ shares of our common stock issuable upon the exercise of outstanding warrants; | |
| ● | ______ shares of our common stock issuable upon the exercise of outstanding options; and | |
| ● | ______ shares of our common stock issuable upon the conversion of notes and other evidence of indebtedness. |
| 10 |
SUMMARY FINANCIAL DATA
The following tables summarize our financial data for the periods presented and should be read together with the sections of this prospectus entitled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and our financial statements and related notes thereto appearing elsewhere in this prospectus. The following summary statements of operations data for the years ended December 31, 2018 and 2017 and the nine-months ended September 30, 2019 and 2018 have been derived from our financial statements and footnotes included elsewhere in this prospectus. Our historical results are not necessarily indicative of the results we expect in the future.
| Operating Summary | Year ended December 31, 2018 | Year ended December 31, 2017 | Nine-months ended September 30, 2019 (unaudited) | Nine-months ended September 30, 2018 (unaudited) | ||||||||||||
| Revenues, net | $ | 1,510,875 | $ | 610,323 | $ | 439,505 | $ | 1,134,899 | ||||||||
| Cost of Goods Sold | (699,852 | ) | (274,707 | ) | (254,479 | ) | (521,353 | ) | ||||||||
| Gross Profit | 811,023 | 335,616 | 185,026 | 613,546 | ||||||||||||
| Operating Expenses | (4,833,518 | ) | (2,337,886 | ) | (3,540,412 | ) | (3,689,560 | ) | ||||||||
| Net Operating Loss | (4,022,495 | ) | (2,002,270 | ) | (3,355,386 | ) | (3,076,014 | ) | ||||||||
| Other Income (Expense) | (1,727 | ) | 17,036 | (295,354 | ) | (859 | ) | |||||||||
| Net Loss | $ | (4,024,222 | ) | $ | (1,985,234 | ) | $ | (3,650,740 | ) | $ | (3,076,873 | ) | ||||
The following table sets forth:
| ● | our summary balance sheet data as of September 30, 2019; and | |
| ● | our pro forma summary balance sheet data assuming this offering was effective on September 30, 2019. |
The pro forma summary balance sheet data is for informational purposes only and does not purport to indicate balance sheet information as of any future date.
| As of September 30, 2019 | ||||||||
| Actual | Pro Forma(1,2) | |||||||
| (Unaudited) | ||||||||
| Balance Sheet data: | ||||||||
| Cash | $ | 7,470 | $ | |||||
| Total assets | 2,114,414 | |||||||
| Total liabilities | 8,379,335 | |||||||
| Accumulated deficit | (65,594,058 | ) | ||||||
| Total stockholders’ equity (deficit) | $ | (6,264,921 | ) | $ | ||||
| (1) | The pro forma information presented above gives effect to the sale of $______ of our common stock in this offering after deducting underwriting discounts and estimated offering fees and expenses of approximately $______ that are payable by us. The pro forma information presented above also gives effect to the automatic conversion of the $815,217 senior convertible note issued July 19, 2019 into shares of our common stock upon the closing of this offering. The pro forma information discussed above is illustrative only and will be adjusted based on the actual public offering price and other terms of this offering determined at pricing. | |
| (2) | The pro forma information presented above does not include convertible notes issued after September 30, 2019 and prior to the effective date of this prospectus in the amount of $423,913, of which $217,391 shall automatically convert into shares of our common stock upon the closing of this offering. |
| 11 |
An investment in our common stock, any warrants to purchase our common stock, or any other security that may be issued by us involves a high degree of risk. You should carefully consider the risks described below, together with all of the other information included elsewhere in this prospectus, before making an investment decision. If any of the following risks actually occur, our business, financial condition, or results of operations could suffer. In that case, the trading price of our shares of our common stock could decline, and you may lose all or part of your investment. You should read the section entitled “Forward-Looking Statements” above for a discussion of what types of statements are forward-looking statements, as well as the significance of such statements in the context of this prospectus.
Our management will have broad discretion as to the use of proceeds from this offering, and we may not use the proceeds effectively.
Our management will have broad discretion in the application of the net proceeds from this offering and could spend the proceeds in ways that do not improve our results of operations or enhance the value of our common stock. You will not have the opportunity, as part of your investment decision, to assess whether these proceeds are being used appropriately. Our failure to apply these funds effectively could have a material adverse effect on our business and cause the price of our common stock to decline.
We are highly dependent on our senior management and certain consultants or other advisors, and if we are not able to retain them or to recruit and retain additional qualified personnel, our business will suffer.
We are highly dependent upon our senior management and certain consultants or other advisors, including David G. Watumull, our President and Chief Executive Officer, David M. Watumull, our Chief Operating Officer, Paresh N. Soni, our Chief Clinical and Regulatory Strategist, Gilbert M. Rishton, our Chief Science Officer, Jon L. Ruckle, our Chief Medical Officer, Timothy J. King, our Vice President, Research, and John B. Russell, our Chief Financial Officer. The loss of services of David G. Watumull or any other member of our senior management could have a material adverse effect on our business, prospects, financial condition, and results of operations. We carry $1 million “key person” life insurance policies on David G. Watumull and David M. Watumull, but we do not carry similar insurance for any of our other senior executives.
We may choose to increase our management personnel. For example, we will need to obtain certain additional functional capability, including regulatory, sales, quality assurance and control, either by hiring additional personnel or by outsourcing these functions to qualified third parties. We may not be able to engage these third parties on terms favorable to us. Also, we may not be able to attract and retain qualified personnel on acceptable terms given the competition for such personnel among companies that operate in our markets. The trend in the pharmaceutical industry of requiring sales and other personnel to enter into non-competition agreements prior to starting employment exacerbates this problem, since personnel who have made such a commitment to their current employers are more difficult to recruit. If we fail to identify, attract, retain, and motivate these highly skilled personnel, or if we lose current employees, our business, prospects, financial conditions, and results of operations could be adversely affected.
Our ability to grow and compete in the future will be adversely affected if adequate capital is not available to us or not available on terms favorable to us.
The ability of our business to grow and compete depends on the availability of adequate capital, which in turn depends in large part on our cash flow from operations and the availability of equity and debt financing. We cannot assure you that our cash flow from operations will be sufficient or that we will be able to obtain equity or debt financing on acceptable terms or at all to implement our growth strategy. As a result, we cannot assure you that adequate capital will be available to finance our current growth plans, take advantage of business opportunities, or respond to competitive pressures, any of which could harm our business. Additionally, if adequate additional financing is not available on acceptable terms, we may not be able to continue our business operations. Any additional capital, investment or financing of our business may result in dilution of our stockholders or be on terms and conditions that impair our ability to profitably conduct our business.
We are dependent upon the success of our products and technologies, which may not be successfully developed or commercialized.
While the FDA does not require clinical trials for dietary supplements, we have conducted and may continue to conduct clinical trials with our dietary supplements to promote scientific and consumer awareness. We also expect to conduct clinical trials with our pharmaceutical candidates. A failure of any clinical trial can occur at any stage of testing. The results of initial clinical testing may not necessarily indicate the results that will be obtained from later or more extensive testing. Additionally, any observations made with respect to blinded clinical data are inherently uncertain as we cannot know which set of data come from subjects treated with active versus placebo. Investors are cautioned not to rely on observations coming from blinded data and not to rely on initial clinical trial results as necessarily indicative of results that will be obtained in subsequent clinical trials or clinical practice.
Additionally, our products are subject to a variety of FDA and other applicable regulatory authorities. The extent of regulations applicable to our products, and the approvals or designations our products may receive from regulatory authorities, such as the FDA, are dependent upon the nature and development of our products and how such products are ultimately commercialized and marketed.
A number of different factors could prevent us from developing or commercializing our products on a timely basis, or at all.
We, the FDA, other applicable regulatory authorities, or an institutional review board (“IRB”), may suspend clinical trials of a product at any time for various reasons, including if we or they believe the subjects participating in such trials are being exposed to unacceptable health risks. Among other reasons, adverse side effects of a product on subjects in a clinical trial could result in the FDA or other regulatory authorities suspending or terminating the trial and refusing to approve or allow continued marketing of a particular product for any or all indications or applications of use.
| 12 |
Clinical trials require the enrollment of a sufficient number of subjects who meet certain eligibility criteria. Rates of subject enrollment are affected by many factors, and delays in subject enrollment can result in increased costs and longer development times.
Clinical trials also require the review and oversight of IRBs, which approve and continually review clinical investigations and protect the rights and welfare of human subjects. An inability or delay in obtaining IRB approval could prevent or delay the initiation and completion of clinical trials, and the FDA may decide not to consider any data or information derived from a clinical investigation not subject to initial and continuing IRB review and approval.
Numerous factors could affect the timing, cost, or outcome of our development and commercialization efforts, including the following:
| ● | delays in filing or acceptance of IND applications for our pharmaceutical candidates; | |
| ● | difficulty in securing centers to conduct clinical trials; | |
| ● | conditions imposed on us by the FDA or other regulatory authorities that are applicable to our business regarding the scope or design of our clinical trials or the method or scope of our sales and marketing practices; | |
| ● | problems in engaging IRBs to oversee trials or problems in obtaining or maintaining IRB approval of studies; | |
| ● | difficulty in enrolling subjects in conformity with required protocols or projected timelines; | |
| ● | third-party contractors failing to comply with regulatory requirements or to meet their contractual obligations to us in a timely manner; | |
| ● | our products having unexpected and different chemical and pharmacological properties in humans than in laboratory testing and interacting with human biological systems in unforeseen, ineffective or harmful ways; | |
| ● | the need to suspend or terminate clinical trials if the subjects are being exposed to unacceptable health risks; | |
| ● | insufficient or inadequate supply or quality of our products or other materials necessary to conduct our clinical trials; | |
| ● | our products not having the desired effects or having undesirable side effects or other unexpected characteristics; | |
| ● | the cost of our clinical trials being greater than we anticipate; | |
| ● | negative or inconclusive results from our clinical trials or the clinical trials of others for similar products or inability to generate statistically significant data confirming the efficacy or safety of the product being tested; | |
| ● | interim or preliminary results of our clinical trials may not be indicative of the final results for such clinical trials or other clinical trials; | |
| ● | interim or preliminary results of our clinical trials do not ensure that the final results such clinical trial or other clinical trials will be positive or statistically significant or clinically meaningful; | |
| ● | results of our clinical trials may not be replicated by other clinical trials; | |
| ● | changes in the FDA’s other applicable regulatory authorities’ requirements for testing during the course of testing; | |
| ● | reallocation of our limited financial and other resources to other programs; and | |
| ● | adverse results obtained by other companies developing similar products. |
It is possible that none of the products we may develop will obtain the appropriate regulatory approvals necessary to begin selling them or that any regulatory approval to market a product may be subject to limitations on the indicated uses for which we may market the product. The time required to obtain FDA and other approvals is unpredictable, but often can take years following the commencement of clinical trials, depending upon the complexity of the product. Any analysis we perform of data from clinical activities is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. Any delay or failure in obtaining required approvals could have a material adverse effect on our ability to generate revenue from the particular product.
We also must comply with clinical trial and post-approval safety and adverse event reporting requirements. Adverse events related to our products must be reported to the FDA in accordance with regulatory timelines based on their severity and expectedness. Failure to make timely safety reports and to establish and maintain related records could result in withdrawal of marketing authorization.
We may also become subject to numerous foreign regulatory requirements governing the conduct of clinical trials, manufacturing and marketing authorization, pricing, and third-party reimbursement. The foreign regulatory approval process includes all of the risks associated with the FDA approval described above as well as risks attributable to the satisfaction of local regulations in foreign jurisdictions. Approval by the FDA does not assure approval by regulatory authorities outside of the United States.
If we fail to comply with FDA regulations our business could suffer.
The manufacture and marketing of pharmaceuticals and dietary supplements are subject to extensive regulation by the FDA and foreign and state regulatory authorities. In the United States, pharmaceutical and dietary supplement companies such as ours must comply with laws and regulations promulgated by the FDA. These laws and regulations require various authorizations prior to a product being marketed in the United States. Manufacturing facilities and practices are also subject to FDA regulations. The FDA regulates the clinical testing, manufacture, labeling, sale, distribution, and promotion of pharmaceuticals and dietary supplements in the United States. Our failure to comply with regulatory requirements, including any future changes to such requirements, could have a material adverse effect on our business, prospects, financial condition, and results of operations.
Even after clearance or approval of a product, we are subject to continuing regulation by the FDA, including the requirements of registering our facilities and listing our products with the FDA. We are subject to reporting regulations. These regulations require us to report to the FDA if any of our products may have caused or contributed to a death or serious injury and such product or a similar product that we market would likely cause or contribute to a death or serious injury. Unless an exemption applies, we must report corrections and removals to the FDA where the correction or removal was initiated to reduce a risk to health posed by the product or to remedy a violation of the Food, Drug, and Cosmetic Act. The FDA also requires that we maintain records of corrections or removals, regardless of whether such corrections and removals are required to be reported to the FDA. In addition, the FDA closely regulates promotion and advertising, and our promotional and advertising activities could come under scrutiny by the FDA.
| 13 |
The FDA also requires that manufacturing be in compliance with its Quality System Regulation, or QSR. The QSR covers the methods and documentation of the design, testing, control, manufacturing, labeling, quality assurance, packaging, storage, and shipping of our products. Our failure to maintain compliance with the QSR requirements could result in the shutdown of, or restrictions on, our manufacturing operations, to the extent we have any, and the recall or seizure of our products, which would have a material adverse effect on our business. In the event that one of our suppliers fails to maintain compliance with our quality requirements, we may have to qualify a new supplier and could experience manufacturing delays as a result.
The FDA has broad enforcement powers. If we violate applicable regulatory requirements, the FDA may bring enforcement actions against us, which could have a material adverse effect on our business, prospects, financial condition, and results of operations. Violations of regulatory requirements, at any stage, including after approval, may result in various adverse consequences, including the delay by a regulatory agency in approving or refusal to approve a product, withdrawal or recall of an approved product from the market, other voluntary agency-initiated action that could delay further development or marketing, as well as the imposition of criminal penalties against the manufacturer and NDA holder.
The extent of FDA regulations applicable to us, and whether our products are ultimately designated as drugs (including active pharmaceutical ingredients) or dietary supplements (including dietary ingredients), will depend upon how our products are ultimately commercialized. Furthermore, our products may be commercialized by us or by other parties through licensing arrangements, joint ventures, or other alliances, and our burden of complying with any regulations applicable to our products will depend upon the nature and extent of any relationships with such partners. While dietary supplements are not as extensively regulated as pharmaceuticals, the extent of any regulations to which we may be subject will depend upon the specific products we ultimately produce.
We have limited experience in managing communications with regulatory authorities, including filing IND applications, filing new drug applications, submitting promotional materials, and generally directing the regulatory processes in all territories.
We may be responsible for managing communications with regulatory authorities, including filing INDs, filing NDAs, submitting promotional materials, and generally directing the regulatory processes in all territories. We have limited experience directing such activities and may not be successful with our planned development strategies, on the planned timelines, or at all. Even if any of our products are designated for “fast track” or “priority review” status or if we seek approval under accelerated approval (Subpart H) regulations, such designation or approval pathway does not necessarily mean a faster development process or regulatory review process or necessarily confer any advantage with respect to approval compared to conventional FDA procedures. Accelerated development and approval procedures will only be available if the indications for which we are developing products remain unmet medical needs and if our clinical trial results support use of surrogate endpoints, respectively. Even if these accelerated development or approval mechanisms are available to us, depending on the results of clinical trials, we may elect to follow the more traditional approval processes for strategic and marketing reasons, since drugs approved under accelerated approval procedures are more likely to be subjected to post-approval requirements for clinical studies to provide confirmatory evidence that the drugs are safe and effective. If we fail to conduct any such required post-approval studies or if the studies fail to verify that any of our products are safe and effective, our FDA approval could be revoked. It can be difficult, time-consuming, and expensive to enroll patients in such clinical trials because physicians and patients are less likely to participate in a clinical trial to receive a drug that is already commercially available. Drugs approved under accelerated approval procedures also require regulatory pre-approval of promotional materials that may delay or otherwise hinder commercialization efforts.
We expect to continue to incur significant research and development expenses, which may make it difficult for us to attain profitability.
We expend substantial funds to develop our products, and additional substantial funds will be required for further research and development, including preclinical and clinical testing, and to manufacture and market any products that are approved for commercial sale. Because the successful development of our products is uncertain, we are unable to precisely estimate the actual funds we will require to develop and potentially commercialize them. In addition, we may not be able to generate enough revenue, even if we are able to commercialize any of our products, to become profitable.
We operate in highly competitive industries, and our failure to compete effectively could adversely affect our market share, financial condition and growth prospects. If competitors are better able to develop and market products that are more effective, or gain greater acceptance in the marketplace than our products, our commercial opportunities may be reduced or eliminated.
The dietary supplement and pharmaceutical industries are constantly evolving, and scientific advances are expected to continue at a rapid pace. This results in intense competition among companies operating in the industry. Other, larger companies may have, or may be developing, products that compete with our products and may significantly limit the market acceptance of our products or render them obsolete. Our technical and/or business competitors would include major pharmaceutical companies, biotechnology companies, consumer health companies, universities, and nonprofit research institutions and foundations. Most of these competitors have significantly greater research and development capabilities than we have, as well as substantial marketing, financial, and managerial resources. ZanthoSyn®, our lead product, primarily competes against products that provide anti-inflammatory health benefits. In addition, there are several other companies, both public and private, that service the same markets as we do, all of which compete to some degree with us.
The primary competitive factors facing us include safety, efficacy, price, quality, breadth of product line, manufacturing quality and capacity, service, marketing, and distribution capabilities. Our current and future competitors may have greater resources, more widely accepted and innovative products and stronger name recognition than we do. Our ability to compete is affected by our ability to:
| ● | develop or acquire new products and innovative technologies; | |
| ● | obtain regulatory clearance and compliance for our products; | |
| ● | manufacture and sell our products cost-effectively; | |
| ● | meet all relevant quality standards for our products in their particular markets; | |
| ● | respond to competitive pressures specific to each of our geographic and product markets; | |
| ● | protect the proprietary technology of our products and avoid infringement of the proprietary rights of others; | |
| ● | market our products; | |
| ● | attract and retain skilled employees, including sales representatives; | |
| ● | maintain and establish distribution relationships; and | |
| ● | engage in acquisitions, joint ventures, or other collaborations. |
| 14 |
Competitors could develop products that are more effective, achieve favorable reimbursement status from third-party payors, cost less, or are ready for commercial introduction before our products. If our competitors are better able to develop and patent products earlier than we can, or develop more effective and/or less expensive products that render our products obsolete or non-competitive, our business will be harmed and our commercial opportunities will be reduced or eliminated.
In addition, competitors and other parties may also seek to impact regulatory status of our products through the filing of citizen petitions or other similar documents.
We believe that the market in which we compete in is also highly sensitive to the introduction of new products, including various prescription drugs, which may rapidly capture a significant share of the market. In the United States, we expect to also compete for sales with heavily advertised national brands manufactured by large pharmaceutical, biotechnology, and consumer health companies, as well as other retailers.
As some products gain market acceptance, we may experience increased competition for those products as more participants enter the market. Currently, we are not a manufacturer. To the extent that we engage third-party manufacturers or use strategic alliances to produce our products, our manufacturing capabilities may not be adequate or sufficient to compete with large scale, direct, or third-party manufacturers. Certain of our potential competitors are larger than us and have longer operating histories, customer bases, greater brand recognition, and greater resources for marketing, advertising, and product promotion. They may be able to secure inventory from vendors on more favorable terms, operate with a lower cost structure, or adopt more aggressive pricing policies. In addition, our potential competitors may be more effective and efficient in introducing new products. We may not be able to compete effectively, and our attempt to do so may require us to increase marketing and/or reduce our prices, which may result in lower margins. Failure to effectively compete could adversely affect our market share, financial condition, and growth prospects.
The pharmaceutical and dietary supplement industries are subject to extensive and complex healthcare regulation. Any determination that we have violated federal or state laws applicable to us that regulate healthcare would have a material adverse effect on our business, prospects, and financial condition.
Federal and state laws regulating healthcare are extensive and complex. The laws applicable to our business are subject to evolving interpretations, and therefore we cannot be sure that a review of our operations by federal or state courts or regulatory authorities will not result in a determination that we have violated one or more provisions of federal or state law. Any such determination could have a material adverse effect on our business, prospects, and financial condition.
Healthcare and insurance legislation may increase the difficulty and cost for us to commercialize our products and affect the prices we may obtain.
The United States and many foreign jurisdictions have enacted or proposed legislative and regulatory changes affecting the healthcare system that could prevent or delay marketing approval of our products, restrict or regulate post-approval activities, and affect our ability to profitably sell any product for which we obtain marketing approval.
In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or Medicare Modernization Act, changed the way Medicare covers and pays for pharmaceuticals. The legislation expanded Medicare coverage for drug purchases by the elderly by establishing Medicare Part D and introduced a new reimbursement methodology based on average sales prices for physician-administered drugs under Medicare Part B. In addition, this legislation provided authority for limiting the number of drugs that Medicare will cover in any therapeutic class under the new Medicare Part D program. Cost reduction initiatives and other provisions of this legislation could decrease the coverage and reimbursement rate that we receive for any of our approved products. While the Medicare Modernization Act applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates. Therefore, any reduction in reimbursement that results from the Medicare Modernization Act may result in a similar reduction in payments from private payors.
In March 2010, former President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or, collectively, the Affordable Care Act, a law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against healthcare fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on pharmaceutical and medical device manufacturers, and impose additional health policy reforms. Among other things, the Affordable Care Act expanded manufacturers’ rebate liability under the Medicaid Drug Rebate Program by increasing the minimum rebate for both branded and generic drugs, effective the first quarter of 2010, and revising the definition of “average manufacturer price,” or AMP, for reporting purposes, which could increase the amount of Medicaid drug rebates manufacturers are required to pay to states. The legislation also extended Medicaid drug rebates, previously due only on fee-for-service utilization, to Medicaid managed care utilization, and created an alternative rebate formula for certain new formulations of certain existing products that is intended to increase the amount of rebates due on those drugs.
The Centers for Medicare and Medicaid Services, which administers the Medicaid Drug Rebate Program, also has proposed to expand Medicaid drug rebates to the utilization that occurs in the United States territories, such as Puerto Rico and the Virgin Islands. Also effective in 2010, the Affordable Care Act expanded the types of entities eligible to receive discounted 340B pricing, although, with the exception of children’s hospitals, these newly eligible entities will not be eligible to receive discounted 340B pricing on orphan drugs. In addition, because 340B pricing is determined based on AMP and Medicaid drug rebate data, the revisions to the Medicaid rebate formula and AMP definition described above could cause the required 340B discounts to increase. Furthermore, as of 2011, the new law imposes a significant annual fee on companies that manufacture or import branded prescription drugs and requires manufacturers to provide a 50% discount off the negotiated price of prescriptions filled by beneficiaries in the Medicare Part D coverage gap, referred to as the “donut hole.” Substantial new provisions affecting compliance have also been enacted, which may affect our business practices with healthcare practitioners. Notably, a significant number of provisions are not yet, or have only recently become, effective. Although it is too early to determine the full effect of the Affordable Care Act, the new law appears likely to continue the downward pressure on pharmaceutical pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs.
| 15 |
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. In August 2011, the former President signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee on Deficit Reduction did not achieve a targeted deficit reduction of at least $1.2 trillion for fiscal years 2012 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year.
We expect that the Affordable Care Act, as well as other healthcare reform measures that have and may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved product, and could seriously harm our future revenues. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our products.
The impact of continued health care reform efforts with respect to the Affordable Care Act is currently unknown, and may adversely affect our business model.
Since its enactment, there have been judicial and Congressional challenges to numerous provisions of the Affordable Care Act. In January 2017, Congress voted to adopt a budget resolution for fiscal year 2017, or the Budget Resolution, that authorizes the implementation of legislation that would repeal portions of the Affordable Care Act. The Budget Resolution is not a law, but it is widely viewed as the first step toward the passage of legislation that would repeal certain aspects of the Affordable Care Act. On January 20, 2017, President Trump signed an Executive Order directing federal agencies with authorities and responsibilities under the Affordable Care Act to waive, defer, grant exemptions from, or delay the implementation of any provision of the Affordable Care Act that would impose a fiscal or regulatory burden on states, individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices. Additionally, on October 12, 2017, President Trump issued another executive order requiring the Secretaries of the Departments of Health and Human Services (“HHS”), Labor, and the Treasury to consider proposing regulations or revising existing guidance to allow more employers to form association health plans that would be allowed to provide coverage across state lines, increase the availability of short-term, limited duration health insurance plans, which are generally not subject to the requirements of the Affordable Care Act, and increase the availability and permitted use of health reimbursement arrangements. On October 13, 2017, the Department of Justice announced that HHS was immediately stopping its cost sharing reduction payments to insurance companies based on the determination that those payments had not been appropriated by Congress. Furthermore, on December 22, 2017, President Trump signed tax reform legislation into law that, in addition to overhauling the federal tax system, also, effective as of January 1, 2019, repeals the penalties associated with the individual mandate. Congress or the President of the United States may also consider subsequent legislation or executive action to replace or eliminate elements of the Affordable Care Act. We will continue to evaluate the effect that the Affordable Care Act and any future measures to modify, repeal or replace the Affordable Care Act have on our business. We are not able to provide any assurance that the continued healthcare reform debate will not result in legislation, regulation, or executive action by the President of the United States that is adverse to our business. We expect continued development in health care reform and cannot provide any assurance that any changes will not be adverse to us our products or strategies.
Orphan drug designation for our products may not confer marketing exclusivity or other expected benefits.
Under the Orphan Drug Act of 1983 (the “Orphan Drug Act”), the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition that (i) affects less than 200,000 persons in the United States, or (ii) affects more than 200,000 in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug for such disease or condition will be recovered from sales in the United States of such drug. The Orphan Drug Act mainly provides incentives intended to make the development of orphan drugs financially viable but does not provide for separate regulatory standards for orphan drugs. Drugs that receive an orphan drug designation do not require prescription drug user fees at the time of marketing application, may qualify the drug development sponsor for certain tax credits, and can be marketed without generic competition for seven years.
We are seeking orphan drug designation for certain products that we believe may qualify for orphan drug designation; however, there can be no assurance that we will request an orphan drug designation for any product, or if requested, that we will receive such orphan drug designation. If we are unable to secure orphan drug designation, our regulatory and commercial prospects may be negatively impacted. Even if we obtain orphan drug designation for a product, we may not be able to obtain marketing approval or maintain orphan drug exclusivity for that product. We may not be the first to obtain marketing approval of any product for which we have obtained orphan drug designation for the orphan-designated indication due to the uncertainties associated with developing pharmaceuticals. In addition, exclusive marketing rights in the United States may be limited if we seek approval for an indication broader than the orphan-designated indication or may be lost if the FDA later determines that the request for designation was materially defective or if we are unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition. Further, even if we obtain orphan drug exclusivity for a product, that exclusivity may not effectively protect the product from competition because different drugs with different active moieties may be approved for the same condition. Even after an orphan drug is approved, the FDA can subsequently approve the same drug with the same active moiety for the same condition if the FDA concludes that the later drug is clinically superior in that it is shown to be safer, more effective, or makes a major contribution to patient care, or the manufacturer of the product with orphan exclusivity is unable to maintain sufficient product quantity. Orphan drug designation may not shorten the development time or regulatory review time of a drug or give the drug any advantage in the regulatory review or approval process, nor does it prevent competitors from obtaining approval of the same drug for indications other than those in which we have been granted orphan drug designation.
| 16 |
If we are unable to obtain and maintain protection of our intellectual property, the value of our products may be adversely affected.
Our business is dependent in part upon our ability to use intellectual property rights to protect our products from competition. To protect our products, we rely on a combination of patent and other intellectual property laws, employment, confidentiality, and invention assignment agreements with our employees and contractors, and confidentiality agreements and protective contractual provisions with our partners, licensors, and other third parties. These methods, however, afford us only limited protection against competition from other products.
We attempt to protect our intellectual property position, in part, by filing patent applications and obtaining patents related to our proprietary technology, inventions, and improvements that are important to our business. However, our patent position is not likely by itself to prevent others from commercializing products that compete directly with our products. Moreover, we do not have patent protection for certain components of our products and our patent applications can be challenged. In addition, we may fail to receive any patent for which we have applied, and any patent owned by us or issued to us could be challenged, invalidated, or held to be unenforceable. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability of a patent, we would lose at least part, and perhaps all, of the patent protection on a product. Even if a defendant does not prevail on a legal assertion of invalidity and/or unenforceability, our patent claims may be construed in a manner that would limit our ability to enforce such claims against the defendant and others.
We also note that any patent granted may not provide a competitive advantage to us. Our competitors may independently develop technologies that are substantially similar or superior to our technologies. Further, third parties may design around our patented or proprietary products and technologies.
We rely on certain trade secrets and we may not be able to adequately protect our trade secrets even with contracts with our personnel and third parties. Also, any third party could independently develop and have the right to use, our trade secret, know-how, and other proprietary information. If we are unable to protect our intellectual property rights, our business, prospects, financial condition, and results of operations could suffer materially.
Our ability to market our products may be impaired by the intellectual property rights of third parties.
Our success depends in part on our products not infringing on the patents and proprietary rights of other parties. For instance, in the United States, patent applications filed in recent years are confidential for 18 months, while older applications are not published until the patent issues. As a result, there may be patents and patent applications of which we are unaware, and avoiding patent infringement may be difficult.
Our industry is characterized by a large number of patents, patent applications, and frequent litigation based on allegations of patent infringement. Competitors may own patents or proprietary rights, or have filed patent applications, related to products that are similar to ours. We may not be aware of all of the patents and pending applications potentially adverse to our interests that may have been issued to others. Moreover, since there may be unpublished patent applications that could result in patents with claims relating to our products, we cannot be sure that our current products will not infringe any patents that might be issued or filed in the future. Based on the litigious nature of our industry and the fact that we may pose a competitive threat to some companies who own or control various patents, we believe it is possible that one or more third parties may assert a patent infringement claim seeking damages or enjoining us from the manufacture or marketing of one or more of our products. Such a lawsuit may have already been filed against us without our knowledge or may be filed in the future. If any future claim of infringement against us was successful, we may be required to pay substantial damages, cease the infringing activity, or obtain the requisite licenses or rights to use the technology, which may not be available to us on acceptable terms, if at all. Even if we were able to obtain rights to a third party’s intellectual property rights, these rights may be non-exclusive, thereby giving our competitors potential access to the same rights and weakening our market position. Moreover, regardless of the outcome, patent litigation could significantly disrupt our business, divert our management’s attention and consume our financial resources. We cannot predict if or when any third-party patent holder will file suit for patent infringement.
We may be involved in lawsuits or proceedings to protect or enforce our intellectual property rights or to defend against infringement claims, which could be expensive and time consuming.
Litigation may be necessary to enforce our intellectual property rights, protect our trade secrets, or determine the validity and scope of the proprietary rights of others. Interference proceedings conducted by a patent and trademark office may be necessary to determine the priority of inventions with respect to our patent applications. Litigation or interference proceedings, including the defense against infringement or invalidity claims, would be expensive and could result in substantial costs and diversion of resources and management attention. In addition, in an infringement proceeding, a court may decide that a patent of ours is not valid or is unenforceable or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology or the product. An adverse determination of any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing. In addition, we may be enjoined from marketing one or more of our products if a court finds that such products infringe the intellectual property rights of a third party.
During litigation, we may not be able to prevent the confidentiality of certain of our proprietary rights because of the substantial amount of discovery required in connection with intellectual property litigation. In addition, during the course of litigation, there could be public announcements of the results of hearings, motions, or other interim proceedings or developments. If investors or customers perceive these results to be negative, it could have a material adverse effect on our business, prospects, financial condition, and results of operations.
Commercialization of our products requires sophisticated sales and marketing teams.
We have limited prior experience with commercializing our products. To successfully continue to commercialize our dietary supplement products and to commercialize any pharmaceutical products, we need to establish and maintain sophisticated sales and marketing teams and/or utilize the resources of any licensee, contractor, or other third party. While we intend to use current Company employees and service providers to lead our marketing efforts, we may choose to expand our marketing and sales team. Experienced sales representatives may be difficult to locate and retain, and all new sales representatives will need to undergo extensive training. There is no assurance that we will be able to recruit and retain sufficiently skilled sales representatives, or that any new sales representatives will ultimately become productive. If we are unable to recruit and retain qualified and productive sales personnel, our ability to commercialize our products and to generate revenues will be impaired, and our business will be harmed.
| 17 |
We have limited experience as a commercial company.
In 2016, we launched our first commercial product, ZanthoSyn®, and we have limited sales to date. As such, we have limited historical financial data upon which to base our projected revenue, planned operating expenses or upon which to evaluate our company and our commercial prospects. Based on our limited experience in developing and marketing new products, we may not be able to effectively:
| ● | drive adoption of our current and future products, including ZanthoSyn®; | |
| ● | attract and retain customers for our products; | |
| ● | provide appropriate levels of customer support for our products; | |
| ● | implement effective marketing strategies to promote awareness of our products; | |
| ● | develop, manufacture, and commercialize new products or achieve an acceptable return on our research and development efforts and expenses; | |
| ● | comply with regulatory requirements applicable to our products; | |
| ● | anticipate and adapt to changes in our market; | |
| ● | maintain and develop strategic relationships with vendors and manufacturers to acquire necessary materials for the production of our existing or future products; | |
| ● | scale our manufacturing activities to meet potential demand at a reasonable cost; | |
| ● | avoid infringement and misappropriation of third-party intellectual property; | |
| ● | obtain any necessary licenses to third-party intellectual property on commercially reasonable terms; | |
| ● | obtain valid and enforceable patents that give us a competitive advantage; | |
| ● | protect our proprietary technology; and | |
| ● | attract, retain, and motivate qualified personnel. |
In addition, a high percentage of our expenses is and will continue to be fixed. Accordingly, if we do not generate revenue as and when anticipated, our losses may be greater than expected and our operating results will suffer.
Market acceptance of ZanthoSyn® and any future products are vital to our future success.
The commercial success of ZanthoSyn® and any future products is dependent upon the acceptance of such products. ZanthoSyn® and any future products may not gain and maintain any significant degree of market acceptance among potential consumers, retailers, healthcare providers, or acceptance by third-party payors, such as health insurance companies. The health applications for ZanthoSyn® and any future products can also be addressed by other products or techniques. The medical community widely accepts alternative treatments, and certain of these other treatments have a long history of use. We cannot be certain that our proposed products and the procedures in which they are used will be able to replace those established treatments or that users will accept and utilize our products or any other medical products that we may market.
Market acceptance will depend upon numerous factors, many of which are not under our control, including:
| ● | the safety and efficacy of our products; | |
| ● | favorable regulatory approval and product labeling; | |
| ● | the availability, safety, efficacy, and ease of use of alternative products or treatments; | |
| ● | our ability to educate potential users on the advantages of our products; | |
| ● | the price of our products relative to alternative technologies; | |
| ● | the availability of third-party reimbursement; and | |
| ● | our distribution channels and any support by retailers. |
If our proposed products do not achieve significant market acceptance, our future revenues and profitability would be adversely affected.
We have limited experience in marketing our products.
We have undertaken limited marketing efforts for ZanthoSyn® and any future products. Our sales and marketing teams compete against the experienced and well-funded sales organizations of competitors. Our future revenues and ability to achieve profitability will depend largely on the effectiveness of our sales and marketing team, and we will face significant challenges and risks related to marketing our services, including, but not limited to, the following:
| ● | the ability of sales representatives to obtain access to or persuade adequate numbers of healthcare providers to recommend and/or purchase and/or use our products; | |
| ● | the ability to recruit, properly motivate, retain, and train adequate numbers of qualified sales and marketing personnel; | |
| ● | the costs associated with hiring, training, maintaining, and expanding an effective sales and marketing team; and | |
| ● | assuring compliance with government regulatory requirements affecting the healthcare industry in general and our products in particular. |
| 18 |
We may seek to establish a network of distributors in selected markets to market, sell, and distribute our products. If we fail to select or use appropriate distributors, or if the sales and marketing strategies of such distributors prove ineffective in generating sales of our products, our future revenues would be adversely affected, and we might never become profitable.
The loss of our largest customer would substantially reduce revenues.
Our customers are material to our success. If we are unable to maintain good relationships with our existing customers, our business could suffer. We sell ZanthoSyn® to GNC under a purchasing agreement. The exclusivity provision under such agreement related to distribution of ZanthoSyn® by GNC in the “brick and mortar” retail channel in the United States expired on October 16, 2019. GNC remains our only distributor of ZanthoSyn® in such channel, but we may expand retail distribution to mass market retailers, other specialty nutrition stores, pharmacies, and other retailers. We cannot provide assurance that GNC will continue to sell ZanthoSyn® at the same levels, or at all.
Commercialization of our products requires sophisticated sales and marketing teams.
We have limited prior experience with commercializing our products. To successfully continue to commercialize our dietary supplement products and to commercialize any pharmaceutical products, we need to establish and maintain sophisticated sales and marketing teams and/or utilize the resources of any licensee, contractor, or other third party. While we intend to use current Company employees and service providers to lead our marketing efforts, we may choose to expand our marketing and sales team. Experienced sales representatives may be difficult to locate and retain, and all new sales representatives will need to undergo extensive training. There is no assurance that we will be able to recruit and retain sufficiently skilled sales representatives, or that any new sales representatives will ultimately become productive. If we are unable to recruit and retain qualified and productive sales personnel, our ability to commercialize our products and to generate revenues will be impaired, and our business will be harmed.
An unexpected interruption or shortage in the supply or significant increase in the cost of components could limit our ability to manufacture any products, which could reduce our sales and margins.
To the extent we engage in relationships with contract manufacturers in the future, an unexpected interruption of supply or a significant increase in the cost of components, whether to us or to our contract manufacturers for any reason, such as regulatory requirements, import restrictions, loss of certifications, disruption of distribution channels as a result of weather, terrorism or acts of war, or other events, could result in significant cost increases and/or shortages of our products. Our inability to obtain sufficient amounts of our products or to pass through higher cost of products we offer could have a material adverse effect on our business, financial condition, or results of operations.
We rely on third parties to supply and manufacture our products. If these third parties do not perform as expected or if our agreements with them are terminated, our business, prospects, financial condition, and results of operations would be materially adversely affected.
We outsource our manufacturing to third parties. Our reliance on contract manufacturers and suppliers exposes us to risks, including the following:
| ● | We rely on our suppliers and manufacturers to provide us with the needed products or components in a timely fashion and of an acceptable quality. An uncorrected defect or supplier’s variation in a component could harm our or our third-party manufacturers’ ability to manufacture, and our ability to sell, products and may subject us to product liability claims. | |
| ● | The facilities of our third-party manufacturers must satisfy production and quality standards set by applicable regulatory authorities. Regulatory authorities periodically inspect manufacturing facilities to determine compliance with these standards. If we or our third-party manufacturers fail to satisfy these requirements, the facilities could be shut down. | |
| ● | These manufacturing operations could also be disrupted or delayed by fire, earthquake or other natural disaster, a work stoppage or other labor-related disruption, failure in supply or other logistical channels, electrical outages, or other reasons. If there was any such disruption to any of these manufacturing facilities, our third-party manufacturers would potentially be unable to manufacture our products. | |
| ● | A third-party manufacturer or supplier could decide to terminate our manufacturing or supply arrangement, including due to a disagreement between us and such third-party manufacturer, if the third-party manufacturer determines not to further manufacture our products, or if we fail to comply with our obligations under such arrangements. | |
| ● | If any third-party manufacturer makes improvements in the manufacturing process for our products, we may not own, or may have to share, the intellectual property rights to the innovation. |
We currently rely on a limited number of suppliers to provide key components for our products. If these or other suppliers become unable to provide components in the volumes needed or at an acceptable price or quality, we would have to identify and qualify acceptable replacements from alternative suppliers. We may experience stoppages in the future. We may not be able to find a sufficient alternative supplier in a reasonable time period, or on commercially reasonable terms, if at all, and our ability to produce and supply our products could be impaired.
To the extent we are able to identify alternative suppliers, qualifying suppliers is a lengthy process. There are a limited number of manufacturers and suppliers that may satisfy applicable requirements. In addition, FDA regulations may require additional testing of any components from new suppliers prior to our use of these materials or components, which testing could delay or prevent the supply of components. Moreover, a new manufacturer would have to be educated in, or develop substantially equivalent processes for, production of our products, which could take a significant period of time.
Each of these risks could delay the development or commercialization of our products or result in higher costs or deprive us of potential product revenues. Furthermore, delays or interruptions in the manufacturing process could limit or curtail our ability to meet demand for our products and/or make commercial sales, unless and until the manufacturing capability at the facilities are restored and re-qualified or alternative manufacturing facilities are developed or brought on-line and “scaled up.” Any such delay or interruption could have a material adverse effect on our business, prospects, financial condition, and results of operations.
| 19 |
We may rely on third-party distributors for sales, marketing, and distribution activities.
We may rely on third-party distributors to sell, market, and distribute ZanthoSyn® and any future products. Because we may rely on third-party distributors for sales, marketing, and distribution activities, we may be subject to a number of risks associated with our dependence on these third-party distributors, including:
| ● | lack of day-to-day control over the activities of third-party distributors; | |
| ● | third-party distributors may not fulfill their obligations to us or otherwise meet our expectations; | |
| ● | third-party distributors may terminate their arrangements with us on limited or no notice or may change the terms of these arrangements in a manner unfavorable to us for reasons outside of our control; and | |
| ● | disagreements with our distributors could require or result in costly and time-consuming litigation or arbitration. |
If we fail to establish and maintain satisfactory relationships with third-party distributors, we may be unable to sell, market, and distribute our products, our future revenues and market share may not grow as anticipated, and we could be subject to unexpected costs which would harm our results of operations and financial condition. There is no assurance that our sales through retail stores will be on terms that are favorable to us or at all.
We may not be able to establish or maintain the third-party relationships that are necessary to develop or potentially commercialize some or all of our products.
We expect to depend on collaborators, partners, licensees, contract research organizations, contract manufacturing organizations, clinical research organizations, and other third parties to support our discovery efforts, to manufacture our products and to conduct clinical trials for some or all of our products. We cannot guarantee that we will be able to successfully negotiate agreements for or maintain relationships with collaborators, partners, licensees, contractors, clinical investigators, vendors, and other third parties on favorable terms, if at all. Our ability to successfully negotiate such agreements will depend on, among other things, potential partners’ evaluation of the superiority of our technology over competing technologies, the quality of the preclinical and clinical data that we have generated and the perceived risks specific to developing our products. If we are unable to obtain or maintain these agreements, we may not be able to develop, manufacture, obtain regulatory approvals for, or commercialize our products. We cannot necessarily control the amount or timing of resources that our contract partners will devote to our research and development programs, products or potential products, and we cannot guarantee that these parties will fulfill their obligations to us under these arrangements in a timely fashion. We may not be able to readily terminate any such agreements with contract partners even if such contract partners do not fulfill their obligations to us. We may experience stoppages in the future. We may not be able to find a sufficient alternative provider in a reasonable time period, or on commercially reasonable terms, if at all, and our ability to produce and supply our products could be impaired.
We do not intend to pay dividends on our common stock so any returns will be limited to the value of our stock.
We have never declared or paid any cash dividends on our common stock or preferred stock. We currently anticipate that we will retain future earnings for the development, operation, and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. Any return to common or preferred stockholders will therefore be limited to the appreciation of their stock.
We have a history of operating losses, have received a going concern opinion from our auditors, and may not have sufficient funds to complete the development and commercialization of our pharmaceutical candidates.
We have incurred substantial net losses since our inception and may continue to incur losses for the foreseeable future, as we continue our product development activities. As a result of our limited operating history, we have limited historical financial data that can be used in evaluating our business and our prospects and in projecting our future operating results. Through September 30, 2019, we have accumulated a total deficit of $65,594,058.
Additionally, we have received a “going concern” opinion from our independent registered public accounting firm. We expect that our marketing program for ZanthoSyn® will continue to focus on outreach to physicians, healthcare professionals, retail personnel, and consumers, and anticipate further losses in the development of our consumer business. We also plan to advance the research and development of our pharmaceutical candidates and anticipate further losses in the development of our pharmaceutical business. As a result of these and other factors, management has determined there is substantial doubt about the Company’s ability to continue as a going concern. Our ability to continue as a going concern is dependent upon our ability to raise additional capital and implement our business plan. If we are unable to achieve or sustain profitability or to secure additional financing on acceptable terms, we may not be able to meet our obligations as they come due, raising substantial doubts as to our ability to continue as a going concern. Any such inability to continue as a going concern may result in our common stock holders losing their entire investment. There is no guarantee that we will become profitable or secure additional financing on acceptable terms. Our consolidated financial statements contemplate that we will continue as a going concern and do not contain any adjustments that might result if we were unable to continue as a going concern. Changes in our operating plans, our existing and anticipated working capital needs, the acceleration or modification of our expansion plans, increased expenses, potential acquisitions or other events will all affect our ability to continue as a going concern.
We expect to use the net proceeds of this offering to fund our research, development, and clinical programs, including the development of our pharmaceutical candidates and for other general corporate purposes. We expect to continue to operate our business at a net loss and will need additional investment to complete such research, development and clinical programs and to commercialize any products that may be successfully developed. We cannot provide assurance that such additional investment or other sources of funding would be available on acceptable terms or at all.
| 20 |
Our Reverse Stock Split may not result in a proportional increase in the per share price of our common stock
The effect of the Reverse Stock Split on the market price for our common stock cannot be accurately predicted. In particular, we cannot assure you that the prices for shares of the common stock after the Reverse Stock Split will increase proportionately to prices for shares of our common stock immediately before the Reverse Stock Split. The market price of our common stock may also be affected by other factors which may be unrelated to the Reverse Stock Split or the number of shares outstanding.
Furthermore, even if the market price of our common stock does rise following the Reverse Stock Split, we cannot assure you that the market price of our common stock immediately after the proposed Reverse Stock Split will be maintained for any period of time. Moreover, because some investors may view the Reverse Stock Split negatively, we cannot assure you that the Reverse Stock Split will not adversely impact the market price of our common stock. Accordingly, our total market capitalization after the Reverse Stock Split may be lower than the market capitalization before the Reverse Stock Split.
Investors in this offering will experience immediate and substantial dilution in net tangible book value.
The public offering price will be substantially higher than the net tangible book value per share of our outstanding shares of our common stock. As a result, investors in this offering will incur immediate dilution of $______ per share, based on the assumed public offering price of $______ per share. Investors in this offering will pay a price per share that substantially exceeds the book value of our assets after subtracting our liabilities. See “Dilution” for a more complete description of how the value of your investment will be diluted upon the completion of this offering.
We have outstanding securities and the warrants issued in connection with this offering provide for recognition of derivative liabilities under U.S. Generally Accepted Accounting Principles (“U.S. GAAP”), which in the future may become significant and would reduce our shareholders’ equity balance that results in adverse consequences.
We have securities that are issued and outstanding after the closing of this offering, and the warrants issued to investors and our underwriters in this offering, require us to recognize derivative liabilities under U.S. GAAP. This derivative liability is described in the footnotes to our financial statements that included in this prospectus, including footnote 2 and footnote 9 to our financial statements as of and for the period ending September 30, 2019. The amount of this derivative liability as of September 30, 2019 was $246,414. The amount of this liability is determined as of each reporting date (that is, the end of each fiscal quarter). Because we estimate the fair value of the conversion feature in its issued convertible note as derivative financial instrument at issuance and at each subsequent reporting date using the Black-Scholes valuation model, the amount of this derivative liability may increase during future reporting periods based on the components of such valuation model, which we do not control, and the amount of the derivative liability may be material in future periods. An increase in this derivative liability results in a decrease of our stockholders’ equity. Any such decrease in our stockholders’ equity resulting from an increase of derivative liabilities may adversely impact our ability to maintain the minimum amount of stockholders’ liability required for us to maintain the listing of our common stock on the Nasdaq Capital Market.
We may be subject to product liability claims. Our insurance may not be sufficient to cover these claims, or we may be required to recall our products.
Our business is to develop and commercialize, among other things, pharmaceuticals and dietary supplements. As a result, we will face an inherent risk of product liability claims. The pharmaceutical and dietary supplement industries have been historically litigious. Since our products are to be used in the human body, manufacturing errors, design defects, or packaging defects could result in injury or death to the patient or consumer. This could result in a recall of one or more of our products and substantial monetary damages. Any product liability claim brought against us, with or without merit, could result in a diversion of our resources, an increase in our product liability insurance premiums, and/or an inability to secure coverage in the future. We may also have to pay any amount awarded by a court in excess of our policy limits. In addition, any recall of our products, whether initiated by us or by a regulatory authority, may result in adverse publicity for us that could have a material adverse effect on our business, prospects, financial condition, and results of operations. Our product liability insurance policies have various exclusions; therefore, we may be subject to a product liability claim or recall for which we have no insurance coverage. In such a case, we may have to pay the entire amount of the award or costs of the recall. Finally, product liability insurance supplements or renewals may be expensive and may not be available in the future on acceptable terms, or at all.
If we experience product recalls, we may incur significant and unexpected costs and damage to our reputation and, therefore, could have a material adverse effect on our business, financial condition, or results of operations.
We may be subject to product recalls, withdrawals, or seizures if any of our products are believed to cause injury or illness or if we are alleged to have violated governmental regulations in the manufacture, labeling, promotion, sale, or distribution of our products. A recall, withdrawal, or seizure of any of our products could materially and adversely affect consumer confidence in our brands and lead to decreased demand for our products. In addition, a recall, withdrawal, or seizure of any of our products would require significant management attention, would likely result in substantial and unexpected expenditures and could materially and adversely affect our business, financial condition, or results of operations.
Our insurance liability coverage is limited and may not be adequate to cover potential losses.
In the ordinary course of business, we purchase insurance coverage (e.g., liability coverage) to protect us against claims made by third parties and employees for product liability, property damage, or personal injuries. However, the protection provided by such insurance is limited in significant respects and, in some instances, we have no coverage and certain of our insurance policies have substantial “deductibles” or have limits on the maximum amounts that may be recovered. Insurers also have exclusions or limitations of coverage for claims related to certain perils including, but not limited to, product liability, mold, and terrorism. If a series of losses occurred, such as from a series of lawsuits, each of which were subject to the deductible amount, or if the maximum limit of the available insurance was substantially exceeded, we could incur losses in amounts that would have a material adverse effect on our results of operations and financial condition.
| 21 |
If we are unable to manage our expected growth, our future revenue and operating results may be adversely affected.
Our anticipated growth is expected to place a significant strain on our management, operational and financial resources. Our current and planned personnel, systems, procedures and controls may not be adequate to support our anticipated growth. To manage our growth, we will be required to improve existing, and implement new, operational and financial systems, procedures, and controls, and to expand, train, and manage our growing employee base. We expect that we may need to increase our management personnel to oversee our expanding operations. Recruiting and retaining qualified individuals can be difficult. If we are unable to manage our growth effectively, or are unsuccessful in recruiting qualified management personnel, our business, prospects, financial condition, and results of operations could be harmed.
An active, liquid, and orderly market for our common stock or Purchase Warrants may not develop.
Our common stock is expected to trade on Nasdaq as and we intend to seek a listing on Nasdaq for our Purchase Warrants. An active trading market for our common stock or Purchase Warrants may never develop or be sustained. If an active market for our common stock or Purchase Warrants does not continue to develop or is not sustained, it may be difficult for investors to sell shares of our common stock or Purchase Warrants without depressing the market price and investors may not be able to sell the shares of our common stock or Purchase Warrants at all. An inactive market may also impair our ability to raise capital by selling our common stock or Purchase Warrants and may impair our ability to acquire other businesses, applications, or technologies using our common stock or Purchase Warrants as consideration, which, in turn, could materially adversely affect our business.
While we are seeking to list our Purchase Warrants on Nasdaq, there is no assurance that our Purchase Warrants will be listed on Nasdaq or any stock exchange.
While we are seeking to list our Purchase Warrants on Nasdaq, we cannot ensure that our Purchase Warrants will be accepted for listing on Nasdaq or any exchange. Should our Purchase Warrants be rejected for listing, we will seek to have our Purchase Warrants quoted on the OTC Markets, in which event the trading price of our Purchase Warrants could suffer, the trading market for our Purchase Warrants may be less liquid, and our Purchase Warrant price may be subject to increased volatility. If we fail to list our Purchase Warrants on the OTC Markets, there will be no public market for our Purchase Warrants.
The Purchase Warrants may not have any value.
Each Purchase Warrant will have an exercise price of not less than 100% of the last reported sale price of our common stock as of the close of the trading day immediately preceding the pricing of this offering and will expire on the fifth anniversary of the date they first become exercisable. In the event our common stock price does not exceed the exercise price of the Purchase Warrants during the period when the warrants are exercisable, the Purchase Warrants may not have any value.
We have a substantial number of outstanding options, warrants and other convertible securities which may cause significant dilution in our shares of common stock.
As described in “Description of Securities – Options”, “Description of Securities – Warrants” and “Description of Securities – Convertible Promissory Notes”, we have a substantial number of outstanding options, warrants, and convertible notes that are outstanding. As described in such section, certain of these securities include anti-dilution provisions which reduces the exercise or conversion price per share of our common stock if we issue shares of common stock at a price that is lower than the then exercise or conversion price. These options, warrants, convertible securities and the Purchase Warrants that are being issued in this offering provide the right to purchase additional shares of common stock at a price that may be less than the then prevailing market price per share of common stock which may cause significant dilution of our shares of common stock. Further, the anti-dilution provisions may adversely affect our ability to raise additional equity capital.
The market price of our common stock may be volatile and may be affected by market conditions beyond our control, and you may not be able to sell our securities.
Companies trading in the stock market in general have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our securities, regardless of our actual operating performance.
The market for our common shares is characterized by significant price volatility when compared to seasoned issuers, and we expect that our share price will continue to be more volatile than a seasoned issuer for the indefinite future. The volatility in our share price is attributable to a number of factors. First, our shares of common stock are sporadically and thinly traded. As a consequence of this lack of liquidity, the trading of relatively small quantities of shares by our stockholders may disproportionately influence the price of those shares in either direction. The price for our shares could, for example, decline precipitously in the event that a large number of shares of our common stock are sold on the market without commensurate demand, as compared to a seasoned issuer which could better absorb those sales without adverse impact on its share price. Second, we are a speculative or “risky” investment due to our limited operating history and lack of profits to date, and uncertainty of future market acceptance for our potential products. As a consequence of this enhanced risk, more risk-averse investors may, under the fear of losing all or most of their investment in the event of negative news or lack of progress, be more inclined to sell their shares on the market more quickly and at greater discounts than would be the case with the stock of a seasoned issuer. Many of these factors are beyond our control and may decrease the market price of our common stock, regardless of our operating performance. We cannot make any predictions or projections as to what the prevailing market price for our common stock will be at any time, including as to whether our common stock will sustain its current market price, or as to what effect the sale of shares or the availability of common stock for sale at any time will have on the prevailing market price.
| 22 |
The market price of our common stock is subject to significant fluctuations in response to, among other factors:
| ● | The significant downward pressure on our common stock price caused by the sale of a significant number of shares could cause our common stock price to decline, thus allowing short sellers of our common stock an opportunity to take advantage of any decrease in the value of our common stock; | |
| ● | The presence and action of short sellers in our common stock; | |
| ● | market acceptance of our existing products, as well as products in development; | |
| ● | the timing of regulatory approvals; | |
| ● | our ability or the ability of third-party distributors to sell, market, and distribute our products; | |
| ● | our ability or the ability of our contract manufacturers to manufacture our products efficiently; | |
| ● | changes in our financial performance or a change in financial estimates or recommendations by securities analysts; | |
| ● | our ability to raise additional funds to complete development of our pharmaceutical product candidates; | |
| ● | announcements of innovations or new products or services by us or our competitors; | |
| ● | the emergence of new competitors or success of our existing competitors; | |
| ● | operating and market price performance of other companies that investors deem comparable; | |
| ● | sales or purchases of our common stock by insiders; | |
| ● | commencement of, or involvement in, litigation; | |
| ● | changes in governmental regulations; and | |
| ● | general economic conditions and slow or negative growth of related markets. |
In addition, if the market for stock in our industry, or the stock market in general, experiences a loss of investor confidence, the market price of our common stock could decline for reasons unrelated to our business, financial condition or results of operations.
If any of the foregoing occurs, it could cause the price of our common stock to fall and may expose us to lawsuits that, even if unsuccessful, could be costly to defend and distract our Board of Directors and management.
We could be subject to securities class action litigation following a market price decline of our common stock.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because pharmaceutical companies have experienced significant stock price volatility in recent years. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
We are currently subject to penny stock regulations and restrictions and if we continue to be subject to such regulations and restrictions you may have difficulty selling shares of our common stock.
The Commission has adopted regulations which generally define so-called “penny stocks” as an equity security that has a market price of less than $5.00 per share or an exercise price of less than $5.00 per share, subject to certain exemptions. Our common stock is a “penny stock”, and we are subject to Rule 15g-9 under the Securities and Exchange Act of 1934, as amended (the “Exchange Act”), or the Penny Stock Rule. This rule imposes additional sales practice requirements on broker-dealers that sell such securities to persons other than established customers and “accredited investors” (generally, individuals with a net worth in excess of $1,000,000 or annual income exceeding $200,000, or $300,000 together with their spouses). For transactions covered by Rule 15g-9, a broker-dealer must make a special suitability determination for the purchaser and receive the purchaser’s written consent to the transaction prior to sale. As a result, this rule affects the ability of broker-dealers to sell our securities and affects the ability of purchasers to sell any of our securities in the secondary market.
For any transaction involving a penny stock, unless exempt, the rules require delivery, prior to any transaction in a penny stock, of a disclosure schedule prepared by the Commission relating to the penny stock market. Disclosure is also required to be made about sales commissions payable to both the broker-dealer and the registered representative and current quotations for the securities. Finally, monthly statements are required to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stock.
After giving effect to this Offering and the Reverse Stock Split and our listing on the Nasdaq Capital Market, our common stock will not be a “penny stock”. There can be no assurance that our shares of common stock will continue to not be a “penny stock” because of its price or qualification for exemption from the Penny Stock Rule. In any event, even if our common stock were exempt from the Penny Stock Rule, we would remain subject to Section 15(b)(6) of the Exchange Act, which gives the Commission the authority to restrict any person from participating in a distribution of penny stock if the Commission finds that such a restriction would be in the public interest.
In addition to the “penny stock” rules described above, the Financial Industry Regulatory Authority (“FINRA”) has adopted similar rules that may also limit a stockholder’s ability to buy and sell our common stock. FINRA rules require that in recommending an investment to a customer, a broker-dealer must have reasonable grounds for believing that the investment is suitable for such customer. Prior to recommending speculative low priced securities to their non-institutional customers, broker-dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status, investment objectives and other information. Under interpretations of these rules, FINRA believes that there is a high probability that speculative low priced securities will not be suitable for at least some customers. The FINRA requirements make it more difficult for broker-dealers to recommend that their customers buy our common stock, which may limit your ability to buy and sell our stock and have an adverse effect on the market for our shares.
| 23 |
We may issue shares of preferred stock that subordinate your rights and dilute your equity interests.
We believe that for us to successfully execute our business strategy we will need to raise investment capital and it may be preferable or necessary to issue preferred stock to investors. Preferred stock may grant the holders certain preferential rights in voting, dividends, liquidation, or other rights in preference over a company’s common stock.
The issuance by us of preferred stock could dilute both the equity interests and the earnings per share of existing holders of our common stock. Such dilution may be substantial, depending upon the number of shares issued. The newly authorized shares of preferred stock could also have voting rights superior to our common stock, and in such event, would have a dilutive effect on the voting power of our existing stockholders.
Any issuance of preferred stock with voting rights could, under certain circumstances, have the effect of delaying or preventing a change in control of us by increasing the number of outstanding shares entitled to vote and by increasing the number of votes required to approve a change in control of us. Shares of voting or convertible preferred stock could be issued, or rights to purchase such shares could be issued, to render more difficult or discourage an attempt to obtain control of us by means of a tender offer, proxy contest, merger or otherwise. Such issuances could therefore deprive our stockholders of benefits that could result from such an attempt, such as the realization of a premium over the market price that such an attempt could cause. Moreover, the issuance of such shares of preferred stock to persons friendly to our Board of Directors could make it more difficult to remove incumbent managers and directors from office even if such change were to be favorable to stockholders generally.
Provisions in our corporate charter documents and under Delaware law could make an acquisition of us more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our corporate charter and our bylaws may discourage, delay, or prevent a merger, acquisition, or other change in control of us that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our Board of Directors. Because our Board of Directors is responsible for appointing the members of our management team, these provisions could in turn affect any attempt by our stockholders to replace current members of our management team. Among others, these provisions include the following:
| ● | our Board of Directors will have the right to elect directors to fill a vacancy created by the expansion of our Board of Directors or the resignation, death, or removal of a director, which will prevent stockholders from being able to fill vacancies on our Board of Directors; | |
| ● | our stockholders will not be able to act by written consent or call special stockholders’ meetings; as a result, a holder, or holders, controlling a majority of our capital stock would not be able to take certain actions other than at annual stockholders’ meetings or special stockholders’ meetings called by our Board of Directors, the chairman of our board, the chief executive officer, or the president; | |
| ● | our certificate of incorporation will prohibit cumulative voting in the election of directors, which limits the ability of minority stockholders to elect director candidates; | |
| ● | our stockholders will be required to provide advance notice and additional disclosures in order to nominate individuals for election to our Board of Directors or to propose matters that can be acted upon at a stockholders’ meeting, which may discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise attempting to obtain control of our company; and | |
| ● | our Board of Directors will be able to issue, without stockholder approval, shares of undesignated preferred stock, which makes it possible for our Board of Directors to issue preferred stock with voting or other rights or preferences that could impede the success of any attempt to acquire us. |
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
We cannot predict the effect the recent U.S. tax reform will have on us.
On December 22, 2017, President Trump signed the Tax Act into law, resulting in sweeping changes to the tax code. The Tax Act, inter alia, reduced the corporate tax rate to 21%, reduced interest expense deductibility, increased capitalization amounts for deferred acquisition costs, eliminated the corporate alternative minimum tax, and reduced the dividend received deduction. Most of the changes in the Tax Act are effective as of January 1, 2018. We are currently unable to predict whether this legislation would have a cumulative positive or negative impact on us.
The terms of this offering may limit our ability to use and benefit from our net operating losses to offset any future taxable income.
Our net operating losses (“NOLs”) may be available to offset our future taxable income to the extent permitted under the Internal Revenue Code (the “IRC”). Under IRC Section 382, the use of NOL carryforwards, capital loss carryforwards, and other tax credit carryforwards may be significantly limited if a change in ownership of a company occurs. A change in ownership under IRC Section 382 is defined, generally, as a cumulative change of 50 percentage points or more in the ownership positions of certain stockholders owning 5% or more of a company’s common stock over a three-year rolling period. If we were to have a change of ownership within the meaning of IRC Section 382, then under certain conditions, our annual federal NOL utilization could be limited to an amount equal to our market capitalization (valued at the time of the ownership change) multiplied by the federal long-term tax exempt rate.
US trade policy could adversely affect our costs.
The future of U.S. trade policies is not certain and has had an effect on the global economy as well as our ability to source product or components from certain countries. The response to current U.S. trade policy by sovereign nations is dynamic and cannot be predicted by us. Other nations may reciprocate in trade tariffs or take other actions that could have an adverse effect on the U.S. economy in general and our ability to acquire raw materials or inventory at acceptable prices.
| 24 |
We estimate the net proceeds to us from the sale of ______ Units at an assumed combined public offering price of $______ per Unit will be approximately $________ after deducting underwriting discounts and estimated offering fees and expenses payable by us. If the underwriters exercise their option to purchase additional shares of our common stock and/or Purchase Warrants in full, we estimate that our net proceeds will be approximately $________ after deducting underwriting discounts and estimated offering fees and expenses payable by us.
We intend to use the net proceeds from the sale of the Units to fund our research, development, and clinical programs, including the funding of our budgeted expenditures to develop our CDX-101 pharmaceutical candidate through IND and to complete our CHASE clinical trial targeting cardiovascular inflammatory health with our ZanthoSyn® astaxanthin dietary supplement, for repayment of approximately $500,000 of current payables owed to our officers or employees, and for other general corporate and working capital purposes, including the repayment of outstanding debt securities as these obligations become due and payable (as described in “Capitalization”), except to the extent that such notes convert into shares of our common stock in accordance with the terms thereof.
The amounts and timing of our use of proceeds will vary depending on a number of factors, including the amount of cash generated or used by our operations. As a result, we will retain broad discretion in the allocation of the net proceeds of this offering. In addition, while we have not entered into any agreements, commitments or understandings relating to any significant transaction as of the date of this prospectus supplement, we may use a portion of the net proceeds to pursue acquisitions, joint ventures, and other strategic transactions. If we obtain additional financing through the issuance of debt or convertible debt securities, then we may use the net proceeds of this offering to repay any such indebtedness.
MARKET PRICE AND DIVIDENDS ON OUR COMMON EQUITY AND RELATED STOCKHOLDER MATTERS
Market Information
Our shares of our common stock are quoted on the OTCQB under the symbol “CDXI.” On the effective date of this prospectus, we expect that trading on Nasdaq will be under the same symbol. The high and low bid quotations for our shares of our common stock for each full quarterly period within the two most recent fiscal years and the first and second fiscal quarters of our current fiscal year are:
The prices set forth below are NOT adjusted for the effect of the Reverse Stock Split.
| Quarter Ended | High | Low | ||||||
| March 31, 2017 | $ | 0.27 | $ | 0.09 | ||||
| June 30, 2017 | $ | 0.23 | $ | 0.12 | ||||
| September 30, 2017 | $ | 0.59 | $ | 0.16 | ||||
| December 31, 2017 | $ | 0.49 | $ | 0.07 | ||||
| March 31, 2018 | $ | 0.44 | $ | 0.13 | ||||
| June 30, 2018 | $ | 0.34 | $ | 0.20 | ||||
| September 30, 2018 | $ | 0.24 | $ | 0.17 | ||||
| December 31, 2018 | $ | 0.22 | $ | 0.17 | ||||
| March 31, 2019 | $ | 0.22 | $ | 0.17 | ||||
| June 30, 2019 | $ | 0.20 | $ | 0.07 | ||||
September 30, 2019 | $ | 0.15 | $ | 0.06 | ||||
Such quotations reflect inter-dealer prices, without retail mark-up, mark-down, or commission and do not necessarily represent actual transactions.
As of November 20, 2019, there were approximately 475 stockholders of record of our common stock. The number of stockholders does not include beneficial owners holding shares through nominee names.
As of November 20, 2019, the last reported sale price of our common stock on the OTCQB was $0.07 per share.
Dividends
We have never paid any cash dividends and intend, for the foreseeable future, to retain any future earnings for the development of our business. Our future dividend policy will be determined by our Board of Directors on the basis of various factors, including our results of operations, financial condition, capital requirements, and investment opportunities.
| 25 |
Securities Authorized for Issuance under Equity Compensation Plans
We adopted, and our stockholders approved, the Cardax, Inc. 2014 Equity Compensation Plan, as amended (the “2014 Plan”), effective as of February 7, 2014. Under such plan, we may grant equity-based incentive awards, including options, restricted stock, and other stock-based awards, to any directors, employees, advisors, and consultants that provide services to us or any of our subsidiaries on terms and conditions that are from time to time determined by us. An aggregate of 50,420,148 shares of our common stock are reserved for issuance under the 2014 Plan (the “Plan Shares”). On December 4, 2018, our stockholders and our Board of Directors authorized the annual increase of the Plan Shares on January 1st of each year, at the discretion of our Board of Directors, by up to such number of shares that is equal to four percent (4%) of the shares of our common stock issued and outstanding as of December 31st of the previous calendar year. Options for the purchase of 45,365,083 shares of our common stock have been granted, options for the purchase of 1,016,357 shares of our common stock have been exercised, and options for the purchase of 3,910,301 shares of our common stock have been forfeited; options for the purchase of 40,438,425 shares of our common stock are outstanding as of the date of this prospectus. In addition, an aggregate of 5,892,667 shares of our common stock have been granted under the 2014 Plan. The purpose of the 2014 Plan is to provide financial incentives for selected directors, employees, advisors, and consultants of Cardax and/or its subsidiaries, thereby promoting the long-term growth and financial success of the Company.
Equity Compensation Plan Information
The following table summarizes information as of the date of this prospectus about our outstanding stock options and shares of our common stock reserved for future issuance under our existing equity compensation plans.
| Plan category | Number of securities to be issued upon exercise of outstanding options, warrants, and rights | Weighted-average exercise price of outstanding options, warrants, and rights | Number of securities remaining available for future issuance under equity compensation plans | |||||||||
| Equity compensation plans approved by security holders | 40,438,425 | $ | 0.40 | 3,256,709 | ||||||||
| Equity compensation plans not approved by security holders | - | - | - | |||||||||
| Total | 40,438,425 | $ | 0.40 | 3,256,709 | ||||||||
Penny Stock Regulations
The Commission has adopted regulations which generally define so-called “penny stocks” as an equity security that has a market price of less than $5.00 per share or an exercise price of less than $5.00 per share, subject to certain exemptions. Our common stock is a “penny stock”, and we are subject to Rule 15g-9 under the Exchange Act, or the Penny Stock Rule. This rule imposes additional sales practice requirements on broker-dealers that sell such securities to persons other than established customers and “accredited investors” (generally, individuals with a net worth in excess of $1,000,000 or annual income exceeding $200,000, or $300,000 together with their spouses). For transactions covered by Rule 15g-9, a broker-dealer must make a special suitability determination for the purchaser and receive the purchaser’s written consent to the transaction prior to sale. As a result, this rule affects the ability of broker-dealers to sell our securities and affects the ability of purchasers to sell any of our securities in the secondary market.
For any transaction involving a penny stock, unless exempt, the rules require delivery, prior to any transaction in a penny stock, of a disclosure schedule prepared by the Commission relating to the penny stock market. Disclosure is also required to be made about sales commissions payable to both the broker-dealer and the registered representative and current quotations for the securities. Finally, monthly statements are required to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stock.
After giving effect to this Offering and the Reverse Stock Split and our listing on the Nasdaq Capital Market, our common stock will not be a “penny stock”. There can be no assurance that our shares of common stock will continue to not be a “penny stock” because of its price or qualification for exemption from the Penny Stock Rule. In any event, even if our common stock were exempt from the Penny Stock Rule, we would remain subject to Section 15(b)(6) of the Exchange Act, which gives the Commission the authority to restrict any person from participating in a distribution of penny stock if the Commission finds that such a restriction would be in the public interest.
In addition to the “penny stock” rules described above, the FINRA has adopted similar rules that may also limit a stockholder’s ability to buy and sell our common stock. FINRA rules require that in recommending an investment to a customer, a broker-dealer must have reasonable grounds for believing that the investment is suitable for such customer. Prior to recommending speculative low priced securities to their non-institutional customers, broker-dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status, investment objectives and other information. Under interpretations of these rules, FINRA believes that there is a high probability that speculative low priced securities will not be suitable for at least some customers. The FINRA requirements make it more difficult for broker-dealers to recommend that their customers buy our common stock, which may limit the ability of our stockholders to sell their shares and have an adverse effect on the market for our shares.
| 26 |
The following table sets forth:
| ● | our capitalization as of September 30, 2019; and | |
| ● | our pro forma capitalization assuming this offering was effective on September 30, 2019. |
Our capitalization following the closing of this offering will be adjusted based on the actual public offering price and other terms of this offering determined at pricing. You should read this information together with our financial statements and the related notes thereto included elsewhere in this prospectus and the information set forth under the headings “Summary Financial Data” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this prospectus.
| As of September 30, 2019 | ||||||||
| Actual | Pro Forma(1,2) | |||||||
| (Unaudited) | ||||||||
| Debt: | ||||||||
| Short-term debt: | ||||||||
| Convertible promissory note, due December 31, 2019 | $ | 150,000 | $ | 150,000 | ||||
| Promissory note, due December 31, 2019 (as amended) | 75,000 | 75,000 | ||||||
| Promissory note, due June 30, 2020 (as amended) | 500,000 | 500,000 | ||||||
| Senior convertible promissory note, due June 30, 2020(3) | 815,217 | - | ||||||
| Convertible promissory notes, due June 30, 2020 | 163,044 | 163,044 | ||||||
| Total short-term debt | 1,703,261 | 888,044 | ||||||
| Long-term debt: | ||||||||
| Revolving inventory financing facility, due January 11, 2022 | 1,000,000 | 1,000,000 | ||||||
| Total long-term debt | 1,000,000 | 1,000,000 | ||||||
| Total debt | 2,703,261 | 1,888,044 | ||||||
| Stockholders’ equity (deficit): | ||||||||
| Preferred stock, $0.001 par value; 50,000,000 shares authorized, 0 shares issued and outstanding | - | - | ||||||
| Common stock, $0.001 par value; 400,000,000 shares authorized, 137,261,594 shares issued and outstanding, actual; 400,000,000 shares authorized, _______ shares issued and outstanding, pro forma(4) | 137,262 | |||||||
| Additional paid-in capital | 59,191,875 | |||||||
| Accumulated deficit | (65,594,058 | ) | (65,594,058 | ) | ||||
| Total stockholders’ equity (deficit) | (6,264,921 | ) | ||||||
| Total capitalization | $ | (3,561,660 | ) | $ | ||||
| (1) | The pro forma information presented above gives effect to the sale of $______ of our common stock in this offering after deducting underwriting discounts and estimated offering fees and expenses of approximately $______ that are payable by us. The pro forma information discussed above is illustrative only and will be adjusted based on the actual public offering price and other terms of this offering determined at pricing. |
| (2) | The pro forma information presented above does not include convertible notes issued after September 30, 2019 and prior to the effective date of this prospectus in the amount of $423,913, of which $217,391 shall automatically convert into shares of our common stock upon the closing of this offering. |
| (3) | The senior convertible note shall, in accordance with its terms, automatically convert into shares of our common stock upon the closing of this offering. |
| (4) | The number of shares of our common stock outstanding immediately after this offering is based on ______ shares of our common stock outstanding as of September 30, 2019, after giving effect to the Reverse Stock Split, and excludes: |
| ● | ______ shares of our common stock issuable upon the exercise of outstanding warrants; | |
| ● | ______ shares of our common stock issuable upon the exercise of outstanding options; and | |
| ● | ______ shares of our common stock issuable upon the conversion of notes and other evidence of indebtedness. | |
Additionally, the table set forth above does not include any securities or transactions after September 30, 2019, including without limitation, the subsequent issuances of our securities that are described in this Registration Statement. | ||
| 27 |
If you invest in our common stock, your interest will be diluted immediately to the extent of the difference between the public offering price per share of our common stock and the as adjusted net tangible book value per share of our common stock immediately after this offering.
As of September 30, 2019, our historical net tangible book value was approximately $(6.7) million or $______ per share of common stock after giving effect to the Reverse Stock Split. Historical net tangible book value per share represents the amount of our total tangible assets less total liabilities, divided by ______, the number of shares of common stock outstanding on September 30, 2019 after giving effect to the Reverse Stock Split.
After giving effect to the Reverse Stock Split and the sale of ______ shares of our common stock in this offering at an assumed public offering price of $______ per share (ascribing no value to the warrants), after deducting underwriting discounts and estimated offering fees and expenses payable by us, our net tangible book value as of September 30, 2019 would have been approximately $______ million, or $______ per share. This amount represents an immediate increase in net tangible book value of $______ per share to our existing stockholders and an immediate dilution in net tangible book value of $______ per share to new investors purchasing shares of our common stock in this offering. We determine dilution by subtracting the net tangible book value per share after the offering from the amount of cash that a new investor paid for a share of common stock.
The following table illustrates this dilution on a per share basis, after giving effect to the Reverse Stock Split:
| Assumed public offering price per share | $ | |||||||
| Historical net tangible book value per share as of September 30, 2019 | $ | |||||||
| Increase in net tangible book value per share attributable to new investors | ||||||||
| Pro forma net tangible book value per share after the offering | ||||||||
| Dilution per share to new investors | $ |
Each $1.00 increase (decrease) in the assumed public offering price of $______ per share would increase (decrease) our pro forma net tangible book value after this offering by approximately $______ million, or approximately $______ per share, and increase (decrease) the dilution per share to new investors by approximately $______ per share, after deducting underwriting discounts and estimated offering fees and expenses payable by us, and assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same. We may also increase or decrease the number of shares we are offering. An increase (decrease) of 100,000 shares in the number of shares offered by us would increase (decrease) our pro forma net tangible book value after this offering by approximately $______ million, or $______ per share, and increase (decrease) the dilution per share to new investors by approximately $______ per share, after deducting underwriting discounts and estimated offering fees and expenses payable by us, and assuming that the assumed public offering price remains the same. The pro forma information discussed above is illustrative only and will be adjusted based on the actual public offering price and other terms of this offering determined at pricing.
If the underwriters exercise their option to purchase additional shares of common stock and/or Purchase Warrants in full, our pro forma net tangible book value per share after this offering would be $______ per share. This amount represents an immediate increase in net tangible book value of $______ per share to our existing stockholders and an immediate dilution in net tangible book value of $______ per share to new investors purchasing shares of our common stock in this offering.
The following table sets forth as of September 30, 2019, on the pro forma basis described above, the differences between the number of shares of common stock purchased from us, the total consideration paid, and the weighted average price per share paid by existing stockholders and by investors purchasing shares of our common stock in this offering, after deducting underwriting discounts and estimated offering fees and expenses, at an assumed public offering price of $______ per share:
| Shares Purchased | Total Consideration | Weighted Average | ||||||||||||||||||
| Number | Percent | Amount | Percent | Price per Share | ||||||||||||||||
| Existing stockholders | (1) | % | $ | 59,329,137 | (2) | % | $ | |||||||||||||
| New investors | % | % | ||||||||||||||||||
| Total | 100.0 | % | $ | 100.0 | % | $ | ||||||||||||||
| (1) | The number of shares of our common stock held by existing stockholders immediately after this offering is based on ______ shares of our common stock outstanding as of September 30, 2019, after giving effect to the Reverse Stock Split, and excludes: |
| ● | ______ shares of our common stock issuable upon the exercise of outstanding warrants; | |
| ● | ______ shares of our common stock issuable upon the exercise of outstanding options; and | |
| ● | ______ shares of our common stock issuable upon the conversion of notes and other evidence of indebtedness. |
| (2) | Represents: | ||||
| Common Stock – par value | $ | ||||
| Additional paid-in capital | |||||
| $ | 59,329,137 | ||||
| 28 |
Management’s Discussion and Analysis
of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of operations in conjunction with our financial statements and notes thereto appearing elsewhere in this prospectus. Our consolidated financial statements are prepared and presented in accordance with generally accepted accounting principles in the United States. In addition to historical financial information, the following discussion and analysis contains forward-looking statements that involve risks, uncertainties, and assumptions. Our actual results could differ materially from those anticipated by these forward-looking statements as a result of many factors. We discuss factors that we believe could cause or contribute to these differences below and elsewhere in this prospectus, including those set forth under “Risk Factors” and “Forward-Looking Statements.”
Corporate Overview and History
We are a development stage biopharmaceutical company primarily focused on the development of pharmaceuticals for chronic diseases driven by inflammation. We also have a commercial business unit that markets dietary supplements for inflammatory health. CDX-101, our astaxanthin pharmaceutical candidate, is being developed for cardiovascular inflammation and dyslipidemia, with a target initial indication of severe hypertriglyceridemia. CDX-301, our zeaxanthin pharmaceutical candidate, is being developed for macular degeneration, with a target initial indication of Stargardt disease. Our pharmaceutical candidates are currently in pre-clinical development, including the planning of IND enabling studies. ZanthoSyn® is a physician recommended astaxanthin dietary supplement for inflammatory health. We sell ZanthoSyn® primarily through wholesale and e-commerce channels. The safety and efficacy of our products have not been directly evaluated in clinical trials or confirmed by the FDA.
At present we are not able to estimate if or when we will be able to generate sustained revenues. Our financial statements have been prepared assuming that we will continue as a going concern; however, given our recurring losses from operations, our independent registered public accounting firm has determined there is substantial doubt about our ability to continue as a going concern. After giving effect to the net proceeds that we will receive from this offering, we expect to have sufficient cash resources to fund our expected operations for at least one year.
Subsequent Events
Convertible Promissory Notes
On November 15, 2019, we entered into a convertible note payable with our Chief Executive Officer, as a lender to the Company, in the amount of $100,000. This note accrues interest payable monthly at the rate of 14% per annum and matures on June 30, 2020. This note and accrued interest thereon may convert into shares of our common stock at $0.10 per share any time at the holder’s option. If this note, or any portion thereof, has not been repaid or converted in full on or prior to the maturity date, then repayment of the unpaid principal balance plus any accrued and unpaid interest thereon, shall be amortized over the following thirty-six (36) months. We have the right to prepay this note without penalty or premium.
On the dates set forth in the table below, we entered into convertible notes payable with lenders, who are also current stockholders, in the amounts set forth in the table below. Each of these notes accrues interest payable monthly at the rate of 8% per annum and matures on June 30, 2020. Each of these notes and accrued interest thereon may convert into shares of our common stock at the conversion price set forth in the table below any time at the holder’s option. If any of these notes, or any portion thereof, has not been repaid or converted in full on or prior to the maturity date, then repayment of the unpaid principal balance plus any accrued and unpaid interest thereon, shall be amortized over the following thirty-six (36) months. We have the right to prepay each of these notes without penalty or premium. Each of these notes were issued with detachable five-year warrants to purchase shares of our common stock as set forth in the table below.
| Issuance Date | Principal Amount | Original Issue Discount | Gross Proceeds | Note Conversion Price Per Share |
Number of Shares Underlying Warrants | Warrant Exercise Price Per Share |
||||||||||||||||||
| October 3, 2019 | $ | 27,174 | $ | 2,174 | $ | 25,000 | $ | 0.12 | 50,000 | $ | 0.12 | |||||||||||||
| October 10, 2019 | 27,174 | 2,174 | 25,000 | 0.12 | 50,000 | 0.12 | ||||||||||||||||||
| October 23, 2019 | 108,696 | 8,696 | 100,000 | 0.12 | 250,000 | 0.15 | ||||||||||||||||||
| 250,000 | 0.20 | |||||||||||||||||||||||
| October 29, 2019 | 27,174 | 2,174 | 25,000 | 0.12 | 50,000 | 0.12 | ||||||||||||||||||
| November 8, 2019 | 16,304 | 1,304 | 15,000 | 0.07 | 30,000 | 0.07 | ||||||||||||||||||
| Total | $ | 206,522 | $ | 16,522 | $ | 190,000 | $ | 0.07-0.12 | 680,000 | $ | 0.07-0.20 | |||||||||||||
On the date set forth in the table below, we entered into a senior convertible note payable with a lender, who is also a current stockholder and beneficial owner of more than 5% of our common stock, in the amount set forth in the table below. This note accrues interest payable monthly at the rate of 8% per annum and matures on June 30, 2020. This note and accrued interest thereon may convert into shares of our common stock at the conversion price then in effect (initially $0.12 per share, subject to adjustment) any time at the holder’s option or automatically upon a qualified financing of at least $5 million at the lower of the conversion price then in effect or a 25% discount to the offering price. The conversion price is subject to adjustment upon the issuance of our common stock or securities convertible into our common stock at a price per share less than the then prevailing conversion price, other than specified exempt issuances; accordingly, on November 8, 2019, the conversion price was adjusted to $0.07 per share. We have the right to prepay this note without penalty or premium. This note was issued with a detachable five-year warrant to purchase shares of our common stock as set forth in the table below. The exercise price of this warrant shall be adjusted in accordance with any adjustment to the conversion price of this note; accordingly, on November 8, 2019, the exercise price was adjusted to $0.07 per share.
| Issuance Date | Principal Amount | Original Issue Discount | Gross Proceeds | Note Conversion Price Per Share | Number of Shares Underlying Warrants | Warrant Exercise Price Per Share | ||||||||||||||||||
| October 16, 2019 | $ | 217,391 | $ | 17,391 | $ | 200,000 | $ | 0.07 | 400,000 | $ | 0.07 | |||||||||||||
General Nutrition Corporation
On October 16, 2019, the exclusivity provision of our purchasing agreement with GNC expired, however, all other provisions of our purchasing agreement with GNC remain in effect. We may expand ZanthoSyn® distribution to mass market retailers, other specialty nutrition stores, pharmacies, and other retailers. We also plan to increase our sales and marketing efforts through e-commerce.
| 29 |
Results of Operations
Results of Operations for the Three and Nine-Months Ended September 30, 2019 and 2018
The following table reflects our operating results for the three and nine-months ended September 30, 2019 and 2018:
| Operating Summary | Three-months ended September 30, 2019 | Three-months ended September 30, 2018 | Nine-months
ended September 30, 2019 | Nine-months
ended September 30, 2018 | ||||||||||||
| Revenues | $ | 229,142 | $ | 549,540 | $ | 439,505 | $ | 1,134,899 | ||||||||
| Cost of Goods Sold | (120,818 | ) | (240,152 | ) | (254,479 | ) | (521,353 | ) | ||||||||
| Gross Profit | 108,324 | 309,388 | 185,026 | 613,546 | ||||||||||||
| Operating Expenses | (1,300,035 | ) | (1,237,019 | ) | (3,540,412 | ) | (3,689,560 | ) | ||||||||
| Net Operating Loss | (1,191,711 | ) | (927,631 | ) | (3,355,386 | ) | (3,076,014 | ) | ||||||||
| Other (Expense) Income | (241,915 | ) | (1,257 | ) | (295,354 | ) | (859 | ) | ||||||||
| Net Loss | $ | (1,433,626 | ) | $ | (928,888 | ) | $ | (3,650,740 | ) | $ | (3,076,873 | ) | ||||
Operating Summary for the Three-Months Ended September 30, 2019 and 2018
Our revenues presently derive from the sale of ZanthoSyn® primarily through wholesale and, to a lesser extent, e-commerce channels. We launched our e-commerce channel in 2016 and began selling to GNC stores in 2017. ZanthoSyn® is currently available at over three thousand GNC corporate stores in the United States. As a result, revenues were $229,142 and $549,540 for the three-months ended September 30, 2019 and 2018, respectively. The decrease in revenues for the three-months ended September 30, 2019 was primarily attributed to decreased replenishment orders by GNC during the current period compared to the previous year. Costs of goods sold were $120,818 and $240,152 for the three-months ended September 30, 2019 and 2018, respectively, and included costs of the product, shipping and handling, sales taxes, merchant fees, and other costs incurred on the sale of goods. Gross profits were $108,324 and $309,388 for the three-months ended September 30, 2019 and 2018, respectively, which represented gross profit margins of approximately 47% and 56%, respectively. The decrease in gross profit margin for the three-months ended September 30, 2019, was primarily attributed to increased promotional activities at GNC stores, which increased the sales discounts passed through to us during the current period.
Operating expenses were $1,300,035 and $1,237,019 for the three-months ended September 30, 2019 and 2018, respectively. Operating expenses primarily consisted of services provided to the Company, including payroll, consultation, and contract services, for research and development, including our clinical trial and pharmaceutical development programs, sales and marketing, and administration. These expenses were paid in accordance with agreements entered with each employee or service provider. Included in operating expenses were $175,712 and $180,562 in stock-based compensation for the three-months ended September 30, 2019 and 2018, respectively. The increase in operating expenses for the period from the same period in the prior year was primarily related to an increase in professional fees as a result of clinical trials and debt and equity issuances and filings.
Other expenses, net, were $241,915 and $1,257 for the three-months ended September 30, 2019 and 2018, respectively. For the three-months ended September 30, 2019, other expenses, net, consisted of the change in the fair value of a derivative liability, loss on abandonment of patents, and interest expense of $20,524, $36,205, and $185,189, respectively. These expenses were partially offset by interest income of $3 realized during the nine-months ended September 30, 2019. For the three-months ended September 30, 2018, other expenses, net, consisted of interest income and interest expense of $7 and $(1,264), respectively.
| 30 |
Operating Summary for the Nine-Months Ended September 30, 2019 and 2018
Our revenues were $439,505 and $1,134,899 for the nine-months ended September 30, 2019 and 2018, respectively. The decrease in revenues for the nine-months ended September 30, 2019 was primarily attributed to a combination of (i) GNC selling through existing ZanthoSyn® inventory we sold to GNC during the prior year, which impacted the timing and amounts of replenishment orders during the current period, (ii) increased promotional activities at GNC stores, which increased the sales discounts passed through to us during the current period, and (iii) GNC inventory adjustments to focus on ZanthoSyn 60 count and 90 count bottles, which are the top performing ZanthoSyn variants at GNC, resulting in a one-time return of remaining ZanthoSyn 30 count bottles from GNC inventory to us. Costs of goods sold were $254,479 and $521,353 for the nine-months ended September 30, 2019 and 2018, respectively, and included costs of the product, shipping and handling, sales taxes, merchant fees, and other costs incurred on the sale of goods. Gross profits were $185,026 and $613,546 for the nine-months ended September 30, 2019 and 2018, respectively, which represented gross profit margins of approximately 42% and 54%, respectively. The decrease in gross profit margin for the nine-months ended September 30, 2019 was primarily attributed to increased promotional activities at GNC stores, which increased the sales discounts passed through to us during the current period.
Operating expenses were $3,540,412 and $3,689,560 for the nine-months ended September 30, 2019 and 2018, respectively. Operating expenses primarily consisted of services provided to the Company, including payroll, consultation, and contract services, for research and development, including our clinical trial and pharmaceutical development programs, sales and marketing, and administration. These expenses were paid in accordance with agreements entered with each employee or service provider. Included in operating expenses were $534,774 and $443,249 in stock-based compensation for the nine-months ended September 30, 2019 and 2018, respectively. The decrease in operating expenses for the period from the same period in the prior year was primarily related to a sales and marketing conference and related expenses that occurred in 2018 but not in 2019.
Other expenses, net, were $295,354 and $859 for the nine-months ended September 30, 2019 and 2018, respectively. For the nine-months ended September 30, 2019, other expenses, net, consisted of the change in the fair value of a derivative liability, loss on abandonment of patents, and interest expense of $3,139, $36,205, and $256,015, respectively. These expenses were partially offset by interest income of $5 realized during the nine-months ended September 30, 2019. For the nine-months ended September 30, 2018, other expenses, net, consisted of interest income of $1,941, other income of $556, and interest expense of $(3,356).
Results of Operations for the Years Ended December 31, 2018 and 2017
The following table reflects our operating results for the years ended December 31, 2018 and 2017:
| Operating Summary | Year
ended December 31, 2018 | Year
ended December 31, 2017 | Change | |||||||||
| Revenues, net | $ | 1,510,875 | $ | 610,323 | $ | 900,552 | ||||||
| Cost of Goods Sold | (699,852 | ) | (274,707 | ) | (425,145 | ) | ||||||
| Gross Profit | 811,023 | 335,616 | 475,407 | |||||||||
| Operating Expenses | (4,833,518 | ) | (2,337,886 | ) | (2,495,632 | ) | ||||||
| Net Operating Loss | (4,022,495 | ) | (2,002,270 | ) | (2,020,225 | ) | ||||||
| Other Income (Expense) | (1,727 | ) | 17,036 | (18,763 | ) | |||||||
| Net Loss | $ | (4,024,222 | ) | $ | (1,985,234 | ) | $ | (2,038,988 | ) | |||
Operating Summary for the Years Ended December 31, 2018 and 2017
Our revenues were $1,510,875 and $610,323 for the years ended December 31, 2018 and 2017, respectively. Cost of goods sold were $699,852 and $274,707 for the years ended December 31, 2018 and 2017, respectively, and included costs of the product, shipping and handling, sales taxes, merchant fees, and other costs incurred on the sale of goods. Gross profits were $811,023 and $335,616 for the years ended December 31, 2018 and 2017, which represented gross profit margins of 54% and 55%, respectively.
Operating expenses were $4,833,518 and $2,337,886, for the years ended December 31, 2018 and 2017, respectively. Operating expenses primarily consisted of services provided to the Company, including payroll and consultation, for research and development, sales and marketing, and administration. These expenses were paid in accordance with agreements entered into with each employee or service provider. Included in operating expenses were $650,271 and $242,146 in stock-based compensation for the years ended December 31, 2018 and 2017, respectively.
Other income (expense) was $(1,727) and $17,036, for the years ended December 31, 2018 and 2017, respectively. For the year ended December 31, 2018, other expense primarily consisted of interest expense of $4,227, which was offset by interest and other income of $2,500. For the year ended December 31, 2017, other income primarily consisted of a State of Hawaii refundable research and development credit of $17,253.
| 31 |
Assets and Liabilities
Assets were $2,114,414 and $2,458,898 as of September 30, 2019 and December 31, 2018, respectively. The decrease was primarily due to a decrease in cash. At September 30, 2019 and December 31, 2018, cash totaled $7,470 and $243,753, respectively. Negative working capital was $5,623,786 and $3,877,290 as of September 30, 2019 and December 31, 2018, respectively, and was primarily due to accrued payroll and paid time off of $3,480,812 and $3,437,011, accrued Board of Director fees and related consultation of $418,546, and accounts payable of $1,706,117 and $1,996,097, less current assets of $1,664,778 and $2,024,364, respectively.
Assets were $2,458,898 and $3,156,685 as of December 31, 2018 and 2017, respectively. The decrease was primarily due to a decrease in cash offset by an increase in inventory. At December 31, 2018 and 2017, cash totaled $243,753 and $2,236,837, respectively. Negative working capital was $3,877,290 and $1,748,373 as of December 31, 2018 and 2017, respectively, and was primarily due to accrued payroll and paid time off of $3,437,011 and $3,404,610, accrued Board of Director fees and related consultation of $418,546, and accounts payable of $1,996,097 and $603,391, less current assets of $2,024,364 and $2,728,174, respectively.
The accrual of payroll and Board of Director fees and related consultation, which occurred from January 2008 to December 2013, was due to significant capital constraints, and was selected in favor of layoffs or furloughs in order to maximize employee and director retention. In 2013 and 2014, the Company initiated repayment on these accrued amounts, utilizing approximately 5% to 10% of proceeds from various financings and plans to continue a structured repayment of the outstanding amounts over time as resources permit.
The issuance of convertible notes resulted in a derivative liability of $246,414 as of September 30, 2019.
Liquidity and Capital Resources
Since our inception, we have sustained operating losses and have used cash raised by issuing securities. We expect to continue to operate with a net loss until we are able to develop and commercialize our pharmaceutical product candidates.
During the nine-months ended September 30, 2019 and 2018, we used cash in operating activities of $3,047,889 and $2,712,155, respectively, and incurred net losses of $3,650,740 and $3,076,873, respectively. During the years ended December 31, 2018 and 2017, we used cash in operating activities of $3,200,528 and $2,080,623, respectively, and incurred net losses of $4,024,222 and $1,985,234, respectively.
As of December 31, 2018, we had a U.S. federal income tax net operating loss (“NOL”) carryforward of approximately $37 million. These NOLs may be available to offset our future taxable income to the extent permitted under the Internal Revenue Code (the “IRC”). Under IRC Section 382, the use of NOL carryforwards, capital loss carryforwards, and other tax credit carryforwards may be significantly limited if a change in ownership of a company occurs. A change in ownership under IRC Section 382 is defined, generally, as a cumulative change of 50 percentage points or more in the ownership positions of certain stockholders owning 5% or more of a company’s common stock over a three-year rolling period. If we were to have a change of ownership within the meaning of IRC Section 382, then under certain conditions, our annual federal NOL utilization could be limited to an amount equal to our market capitalization (valued at the time of the ownership change) multiplied by the federal long-term tax exempt rate.
We require additional financing in order to continue to fund our operations and to pay existing and future liabilities and other obligations.
We intend to use the proceeds from this offering primarily to fund our research, development, and clinical programs, as well as for other general corporate purposes, including working capital and repayment of certain indebtedness (as described in “Use of Proceeds”). After giving effect to the net proceeds that we will receive from this offering, we expect to have sufficient cash resources to fund the budgeted expenditures for our expected operations for at least one year.
We also may obtain additional financing from investors through the private placement of our common stock and warrants to purchase our common stock or through the issuance of debt or convertible debt securities and plan to do so prior to the closing of this offering. There can be no assurance that a financing transaction will be available to us on terms and conditions that we determine are acceptable.
We cannot give any assurance that we will in the future be able to achieve a level of profitability from the sale of existing or future products or otherwise to sustain our operations. These conditions raise substantial doubt about our ability to continue as a going concern. The accompanying financial statements do not include any adjustments to reflect the possible future effects on recoverability and reclassification of assets or the amounts and classification of liabilities that may result from the outcome of this uncertainty.
Any inability to obtain additional financing on acceptable terms will materially and adversely affect us, including requiring us to significantly curtail or cease business operations altogether.
| 32 |
Our working capital and capital requirements at any given time depend upon numerous factors, including, but not limited to:
| ● | revenues from the sale of any products or licenses; | |
| ● | costs of production, marketing and sales capabilities, or other operating expenses; and | |
| ● | costs of research, development, and commercialization of our products and technologies. |
We have undertaken certain actions regarding the advancement of our pharmaceutical development program, the conduct of a dietary supplement clinical trial, and the continued sales and marketing of our commercial dietary supplement. We plan to fund such activities, including compensation to service providers, with a combination of cash and equity payments. The amount of payments in cash and equity will be determined by us from time to time.
We will incur ongoing recurring expenses associated with professional fees for accounting, legal, and other expenses for annual reports, quarterly reports, proxy statements and other filings under the Exchange Act. We estimate that these costs will likely be in excess of $250,000 per year. These obligations will reduce our ability and resources to fund other aspects of our business. We hope to be able to use our status as a public company to increase our ability to use non-cash means of settling obligations and compensate certain independent contractors who provide professional services to us, although there can be no assurances that we will be successful in any of those efforts.
The following is a summary of our cash flows provided by (used in) operating, investing, and provided by financing activities during the periods indicated:
| Cash Flow Summary | Nine-months
ended September 30, 2019 | Nine-months
ended September 30, 2018 | ||||||
| Net Cash from Operating Activities | $ | (3,047,889 | ) | $ | (2,712,155 | ) | ||
| Net Cash from Investing Activities | (58,394 | ) | (30,483 | ) | ||||
| Net Cash from Financing Activities | 2,870,000 | 704,375 | ||||||
| Net Cash Decrease for Period | (236,283 | ) | (2,038,263 | ) | ||||
| Cash at Beginning of Period | 243,753 | 2,236,837 | ||||||
| Cash at End of Period | $ | 7,470 | $ | 198,574 | ||||
Cash Flows from Operating Activities
During the nine-months ended September 30, 2019 and 2018, our operating activities primarily consisted of receipts and receivables from sales, payments or accruals for employees, directors, and consultants for services related to administration, sales and marketing, research and development, and inventory deposits.
Cash Flows from Investing Activities
During the nine-months ended September 30, 2019 and 2018, our investing activities were related to expenditures on patents.
Cash Flows from Financing Activities
During the nine-months ended September 30, 2019, our financing activities consisted of transactions in which we raised proceeds through the issuance of our common stock and convertible and other notes payable. The issuance of the convertible notes resulted in a derivative liability of $246,414 as of September 30, 2019.
The following is a summary of our cash flows provided by (used in) operating, investing, and financing activities during the periods indicated:
| Cash Flow Summary | Year
ended December 31, 2018 | Year
ended December 31, 2017 | ||||||
| Net Cash from Operating Activities | $ | (3,200,528 | ) | $ | (2,080,623 | ) | ||
| Net Cash from Investing Activities | (36,593 | ) | (19,408 | ) | ||||
| Net Cash from Financing Activities | 1,244,037 | 4,178,435 | ||||||
| Net Cash (Decrease) Increase | (1,993,084 | ) | 2,078,404 | |||||
| Cash at Beginning of Year | 2,236,837 | 158,433 | ||||||
| Cash at End of Year | $ | 243,753 | $ | 2,236,837 | ||||
| 33 |
Cash Flows from Operating Activities
During the years ended December 31, 2018 and 2017, our operating activities primarily consisted of receipts and receivables from sales, payments or accruals for employees, directors, and consultants for services related to research and development, sales and marketing, and administration, and deposits for future inventory.
Cash Flows from Investing Activities
During the years ended December 31, 2018 and 2017, our investing activities were primarily related to expenditures on patents.
Cash Flows from Financing Activities
During the years ended December 31, 2018 and 2017, our financing activities primarily consisted of transactions in which we raised proceeds through the issuance of our common stock.
Recently Issued Accounting Pronouncements
In February 2016, the Financial Accountings Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2016-02, Leases. This ASU requires management to recognize lease assets and lease liabilities for all leases. ASU No. 2016-02 retains a distinction between finance leases and operating leases. The classification criteria for distinguishing between finance leases and operating leases are substantially similar to the classification criteria for distinguishing between capital leases and operating leases in the previous lease guidance. The result of retaining a distinction between finance leases and operating leases is that under the lessee accounting model, the effect of leases in the statement of comprehensive income and the statement of cash flows is largely unchanged from previous U.S. GAAP. The guidance in ASU No. 2016-02 is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. The Company applied the modified retrospective approach in adopting this standard. The modified retrospective approach includes a number of optional practical expedients that the Company elected to apply; primarily the identification and classification of leases that commenced before the effective date, initial direct costs for leases that commenced before the effective date, and the ability to use hindsight in evaluating lessee options to extend or terminate a lease or to purchase the underlying asset. As part of this adoption, the Company will, in effect, continue to account for leases that commence before the effective date in accordance with previous U.S. GAAP unless the lease is modified, except that lessees are required to recognize a right-of-use asset and a lease liability for all operating leases at each reporting date based on the present value of the remaining minimum rental payments that were tracked and disclosed under previous U.S. GAAP. This adoption of this standard on January 1, 2019, resulted in the Company recognizing a right-to-use asset and lease liability. The Company elected to not recognize any right-to-use assets or liabilities for leases that are twelve months or less. Lease costs are recognized straight-line over the term of the lease. The adoption of this standard did not impact retained earnings of cash flows of the Company.
In June 2018, the FASB issued ASU No. 2018-07, Compensation – Stock Compensation (Topic 718), Improvements to Nonemployee Share-Based Payment Accounting. This ASU is intended to simplify aspects of share-based compensation issued to non-employees by making the guidance consistent accounting for employee share-based compensation. The guidance in ASU No. 2018-07 is effective for annual reporting periods, and interim periods within those years, beginning after December 15, 2018. The Company is currently in the process of evaluating the impact of the adoption of this ASU on its consolidated financial statements.
In August 2018, the FASB issued ASU No. 2018-13, Fair Value Measurement. This ASU modifies the disclosure requirements on fair value measurements in Topic 820, Fair Value Measurement, based on the concepts in the FASB’s Concepts Statement, including the consideration of costs and benefits. The guidance in ASU No. 2018-13 is effective for annual reporting periods, and interim periods within those years, beginning after December 15, 2019. The Company is currently in the process of evaluating the impact of the adoption of this ASU on its consolidated financial statements.
The Company does not believe that any other recently issued, but not yet effective accounting pronouncements, if adopted, would have a material effect on the consolidated financial statements.
Off-Balance Sheet Arrangements
There are no off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures, or capital resources.
| 34 |
Overview
We are a development stage biopharmaceutical company focused primarily on the development of pharmaceuticals to safely address one of the major underlying causes of many chronic diseases – inflammation – including cardiovascular disease, metabolic disease, liver disease, arthritis, and aging. We also have a commercial business unit that markets dietary supplements for inflammatory health. We believe we are well positioned for growth through the utilization of astaxanthin and zeaxanthin for chronic pharmaceutical applications by safely reducing chronic inflammation at the cellular and mitochondrial level – without inhibiting normal function. Similar mechanisms also support the use of our dietary supplement for inflammatory health.
We believe that our pharmaceutical product candidates and our dietary supplements have competitive advantages primarily relating to a unique combination of the following benefits:
| ● | An excellent safety profile that supports chronic use | |
| ● | Broad anti-inflammatory activity and pleiotropic effects with potential application to several chronic diseases as pharmaceuticals and various areas of health as dietary supplements | |
| ● | Oral dosing convenience | |
| ● | Scalable manufacturing | |
| ● | Economical pricing |
Market Overview
There is broad acceptance in the scientific, medical, and financial communities that chronic inflammation is a significant factor in many chronic diseases, particularly cardiovascular disease. The double-blind, randomized, placebo-controlled CANTOS clinical trial (10,061 patients; Novartis, 2017) and REDUCE-IT clinical trial (8,179 patients; Amarin Corporation, 2018), both published in the New England Journal of Medicine, helped to catalyze and support this consensus. Commonly used anti-inflammatory drugs such as aspirin, ibuprofen, naproxen, COX-2 inhibitors, corticosteroids, and various biologics may reduce inflammation, but they have risks of significant side effects that limit their utility in chronic disease.
We believe that a safe anti-inflammatory is the solution. Our lead pharmaceutical candidate CDX-101, a proprietary prodrug of the naturally occurring marine molecule astaxanthin, may provide the needed combination of an excellent safety profile, anti-inflammatory activity, and economic pricing to become widely used for the prevention and treatment of chronic diseases driven by inflammation.
We are pursuing an initial indication of severe hypertriglyceridemia (triglycerides ≥ 500 mg/dL) for CDX-101. Severe hypertriglyceridemia is associated with chronic inflammation and patients with the disorder have increased cardiovascular disease risk and incidence of pancreatitis. We believe the clinical pathway to FDA drug approval for severe hypertriglyceridemia, which relies on biomarker endpoints (i.e., measuring triglycerides in blood tests over a period of several months), will be more efficient than other potential indications that require clinical outcomes studies (e.g., evaluating heart attacks, strokes, and deaths over a period of several years), and is thus better suited as our initial indication for CDX-101.
An estimated 3.4 million Americans have severe hypertriglyceridemia according to peer-reviewed research published in the American Journal of Cardiology in 2011. Statins, fibrates, and prescription fish oils are all used to manage hypertriglyceridemia. 21% (42 million) of U.S. adults have mixed dyslipidemia (high levels of low-density lipoprotein “LDL” cholesterol with low levels of high-density lipoprotein “HDL” cholesterol and/or high levels of triglycerides), with nearly 6% (11.6 million people) having all three lipid abnormalities. Lovaza, Vascepa, and other prescription fish oils approved for severe hypertriglyceridemia are also used off-label in mixed dyslipidemia patients to reduce moderately elevated triglycerides and aggregate sales of these products for on and off-label use are estimated to be approaching $2 billion annually.
We believe CDX-101 will have several competitive advantages compared to prescription fish oils: (i) ease of administration: oral dosing of large fish oil capsules is problematic, whereas we expect CDX-101 tablets should be far smaller; (ii) scalability: prescription fish oil manufacturing is limited by the declining global fish supply, whereas we believe the synthetic production of CDX-101 is scalable; and (iii) safety: prescription fish oils have certain safety risks, whereas we believe that astaxanthin, the active moiety of CDX-101, has an excellent safety profile.
The REDUCE-IT clinical trial demonstrated that administration of Vascepa resulted in a significant reduction of major adverse cardiovascular events (“MACE”) in patients with mixed dyslipidemia on standard of care, specifically statins, and we believe is the primary basis of Amarin’s request to the FDA to expand Vascepa’s label. The reduction of triglycerides in the REDUCE-IT clinical trial was modest however, and the study’s authors concluded that Vascepa’s ability to reduce other markers of cardiovascular disease, including inflammation and oxidized LDL (as demonstrated in the MARINE and ANCHOR clinical trials), provided the pleiotropic effects that led to reduction of MACE in REDUCE-IT. In human proof-of-concept “pilot” studies conducted by third parties and animal models conducted by third parties and us, astaxanthin, the active moiety of CDX-101, has demonstrated similar pleiotropic effects, which are derived from its broad anti-inflammatory activity, but without the limitations of Vascepa or other prescription fish oils. As a result, we believe this market also presents a major opportunity as a potential second indication for CDX-101.
Beyond cardiovascular disease, we believe CDX-101 could be developed to address other chronic diseases driven by inflammation, including metabolic disease, liver disease, arthritis, and aging, each with potential annual sales exceeding a billion dollars.
We are also developing CDX-301, our zeaxanthin pharmaceutical candidate, for macular degeneration. Our target initial indication for CDX-301 is Stargardt disease, a juvenile form of macular degeneration and potential orphan drug indication. Zeaxanthin has a mechanism of action and excellent safety profile similar to astaxanthin, however, it accumulates in the human eye through uptake by a unique retinal receptor, providing protection against blue light, oxidative damage, and related inflammation that occurs in macular degeneration. Pre-clinical and clinical studies with zeaxanthin have demonstrated proof-of-concept for the treatment of macular disorders. Based on multiple academic and NIH sources, we believe there are no more than 42,000 persons in the United States with Stargardt disease, and therefore we believe a treatment for Stargardt disease may qualify for orphan drug designation. (By statute, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition that affects less than 200,000 persons in the United States.) If CDX-301 receives FDA orphan drug designation for Stargardt disease and obtains FDA drug approval, we expect CDX-301 may benefit from certain advantages as an orphan drug, including orphan drug exclusivity, which means the FDA may not approve any other application, including a full NDA, to market the same drug for the same indication for a period of seven years, except in limited circumstances. We also believe that age related macular degeneration, a larger market estimated to afflict more than three million people in the U.S. alone, presents a major opportunity as a potential second indication for CDX-301. We do not expect to use the proceeds of this offering to pursue the development of CDX-301.
Astaxanthin
Astaxanthin Safety
Astaxanthin is a naturally occurring marine carotenoid found in salmon, microalgae, krill, lobster, and crab. Carotenoids are natural pigments that impart coloration and support animal health and vitality, especially in harsh marine environments. Astaxanthin is responsible for the characteristic red or pink color of salmon and shellfish. Salmon without astaxanthin are smaller, more susceptible to infection, have reproductive problems, and are not strong enough to swim upstream.
| 35 |
Astaxanthin is GRAS as a food substance according to FDA regulations and has undergone extensive toxicity testing by third parties and us with no clinically meaningful issues even at the extremely high doses summarized in the table below:
| Type of Study | Maximum Dosing | |
| Acute Toxicity | >8,000 mg/kg (mouse, rat), 2,000 mg/kg (non-human primates) | |
| Sub-Chronic Toxicity | 1,240 mg/kg (rat), 160 mg/kg (dog) | |
| 1 Year Chronic Toxicity/Carcinogenicity | 1,000 mg/kg (rat), 1,400 mg/kg (mouse), 200 mg/kg (dog) | |
| 2 Year Carcinogenicity | 1,000 mg/kg (rat) | |
| Genotoxicity/Mutagenicity | 2,000 mg/kg (mouse) | |
| Teratogenicity | 1,000 mg/kg (rat), 400 mg/kg (rabbit) |
Commonly used anti-inflammatory drugs such as aspirin, ibuprofen, naproxen, COX-2 inhibitors, corticosteroids, and various biologics have risks of side effects including gastrointestinal bleeding, heart attacks, strokes, and severe infections. Prescription fish oil drugs, while safer than common anti-inflammatory drugs, also have risks of certain side effects. Lovaza and other DHA, EPA combination fish oil drugs, have risks of side effects including back pain, eructation, dysgeusia, and increases in LDL cholesterol. Vascepa has risks of side effects including arthralgia, atrial fibrillation, and increased bleeding. Fenofibrates have risks of side effects including stomach pain, nausea, and back pain.
In contrast, astaxanthin has no known side effects of clinical significance. We believe astaxanthin’s excellent safety profile will be a key competitive advantage compared to other drugs targeting inflammation and lipids.
Astaxanthin Mechanism of Action
The mechanism of action of astaxanthin, the active moiety in CDX-101, is quite different than most drugs, and we believe is responsible for its excellent safety profile. Most drugs target single receptors or enzymes in complex pathways, which can lead to side effects with chronic use. Astaxanthin is distributed systemically, including to the liver and heart, where it localizes in cellular and mitochondrial membranes and reduces the oxidative stress that causes chronic inflammation, without affecting the normal function of inflammatory/metabolic signaling pathways. And unlike other antioxidants such as beta-carotene, Vitamin C, and Vitamin E, astaxanthin spans and stabilizes cellular and mitochondrial membranes (biological lipid bilayers) to function as an aqueous and lipid phase antioxidant without membrane disruption, as proven by X-ray diffraction studies:
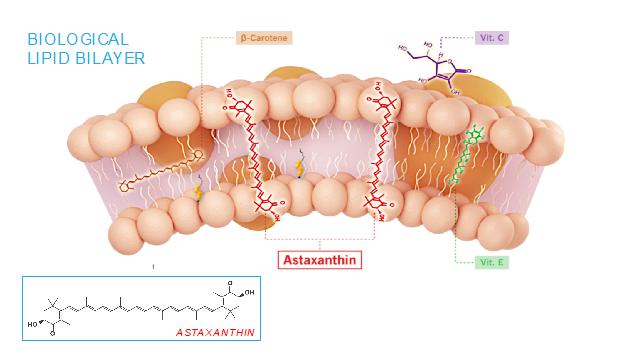
As a result, astaxanthin demonstrates positive and quantifiable pleiotropic effects on many inflammatory cytokines and drug targets.
| 36 |
In human proof-of-concept “pilot” studies conducted by third parties, astaxanthin statistically significantly decreased inflammation and oxidative stress:
| ● | TNF-α decreased (-30%, p=0.0022) | |
| ● | CRP decreased (-20%, p<0.05; two studies) | |
| ● | Oxidative stress decreased (MDA, IsoP, SOD, TAC increased) |
In animal studies conducted by third parties, astaxanthin statistically significantly decreased inflammation and oxidative stress:
| ● | Inflammatory markers decreased in various model systems: |
| ○ | TNF-α, IL-1β, IL-6, CRP, NF-kB, PGE-2, iNOS, MCP-1, MPO, ERK, JNK, COX-2 | |
| ○ | TNF-α decreased equivalent to an equal dose of prednisolone |
| ● | Oxidative stress decreased in mitochondria |
Astaxanthin Research Results
There are more than 2,000 published peer reviewed papers related to astaxanthin, including more than 50 peer reviewed papers published by Cardax and its collaborators (referred to herein as “us”) and more than 50 “pilot” human clinical trials with astaxanthin supplements, more than 20 of which were randomized, double-blind, placebo-controlled human proof-of-concept studies. Highlights of astaxanthin’s pleiotropic effects, which were demonstrated in studies utilizing astaxanthin from natural and synthetic sources, include:
Astaxanthin and Cardiovascular Disease
In human proof-of-concept “pilot” studies conducted by third parties, astaxanthin statistically significantly decreased inflammation, triglycerides, LDL cholesterol, and blood pressure:
| ● | CRP decreased (-20%, p<0.05; two studies) | |
| ● | Triglycerides decreased (-25.8%, p<0.05) | |
| ● | LDL-C decreased (-10.4%, p<0.05) | |
| ● | HDL-C increased (+14.5%, p<0.01) | |
| ● | Apolipoprotein B decreased (-7.5%, p<0.01) | |
| ● | Adiponectin increased (+26%, p<0.01; +14%, p=0.0053; +30%, p=0.01; three studies) | |
| ● | Blood pressure decreased (systolic blood pressure -4.6%, p=0.021; diastolic blood pressure -6.9%, p<0.001; two studies) | |
| ● | Blood flow velocity increased (choroidal, p=0.018, blood transit time, p<0.01) |
In animal studies conducted by third parties and us, astaxanthin demonstrated statistically significant improvements in models of cardiovascular disease:
| ● | CRP and IL-6 decreased | |
| ● | Triglycerides decreased (plasma, hepatic) | |
| ● | Re-thrombosis decreased | |
| ● | Atherosclerosis decreased (aortic arch plaque) | |
| ● | Cholesterol decreased | |
| ● | Blood pressure decreased | |
| ● | Nitric oxide production increased |
Astaxanthin and Metabolic Disease
In human proof-of-concept “pilot” studies conducted by third parties, astaxanthin statistically significantly increased adiponectin and decreased TNF-α and oxidative stress:
| ● | Adiponectin increased (+26%, p<0.01; +14%, p=0.0053; +30%, p=0.01; three studies) | |
| ● | TNF-α decreased (-30%, p=0.0022) | |
| ● | Oxidative stress decreased (MDA, IsoP, SOD, TAC increased) |
In animal studies conducted by third parties, astaxanthin demonstrated statistically significant improvements in models of metabolic disease:
| ● | Fasting blood glucose levels decreased | |
| ● | Insulin levels & sensitivity (HOMA-IR, QUICK) increased | |
| ● | Insulin signaling (PI3K-AKT, IRS-1p) increased | |
| ● | Adiponectin levels increased | |
| ● | Insulin response and glucose tolerance (ipGTT) increased | |
| ● | GLUT-4 translocation increased | |
| ● | JNK, ERK-1 levels decreased | |
| ● | Nitric oxide production increased |
| 37 |
Astaxanthin and Liver Disease
In human proof-of-concept “pilot” studies conducted by third parties, astaxanthin statistically significantly decreased fat accumulation in biopsy-diagnosed NASH patients, decreased TNF-α, improved lipid profile parameters, and decreased oxidative stress:
| ● | NASH disease markers decreased in patients: |
| ○ | Steatosis: p<0.05 | |
| ○ | Nonalcoholic fatty liver disease (“NAFLD”) Activity Score (“NAS”): p<0.08 | |
| ○ | Lobular inflammation decreased: trend |
| ● | TNF-α decreased (-30%, p=0.0022) | |
| ● | Lipid profile parameters improved (LDL, HDL, ApoB, TG) | |
| ● | Oxidative stress decreased (MDA, IsoP, SOD, TAC increased) |
In animal studies conducted by third parties and us, astaxanthin statistically significantly decreased elevated liver enzymes, lipids, insulin resistance, steatosis, and fibrosis:
| ● | Elevated liver enzyme levels decreased | |
| ● | Steatosis decreased | |
| ● | Fibrosis and induced acute hepatitis decreased | |
| ● | Insulin levels & sensitivity (HOMA-IR, QUICK) increased | |
| ● | Insulin signaling (PI3K-AKT, IRS-1p) increased | |
| ● | Adiponectin levels increased |
Astaxanthin and Arthritis
In human proof-of-concept “pilot” non-arthritis studies conducted by third parties, astaxanthin statistically significantly decreased markers of inflammation of relevance to arthritis, including TNF-α and CRP:
| ● | TNF-α decreased (-30%, p=0.0022) | |
| ● | CRP decreased (-20%, p<0.05; two studies) | |
| ● | Adiponectin increased (+26%, p<0.01; +14%, p=0.0053; +30%, p=0.01; three studies) | |
| ● | Oxidative stress decreased (MDA, IsoP, SOD, TAC increased) |
In animal studies conducted by third parties, astaxanthin statistically significantly decreased inflammation, oxidative stress, and joint degeneration:
| ● | Inflammatory markers decreased in various model systems: |
| ○ | TNF-α, IL-1β, IL-6, CRP, NF-kB, PGE-2, iNOS, MCP-1, MPO, ERK, JNK, COX-2 | |
| ○ | TNF-α decreased equivalent to an equal dose of prednisolone |
| ● | Oxidative stress decreased in mitochondria | |
| ● | Cartilage degradation decreased (Mankin score) in surgically-induced model of OA (ACLT, rabbit) |
Astaxanthin and Aging
In human studies conducted by third parties, activation of the FOXO3 gene has been linked to decreased inflammation and aging.
| ● | Activation of anti-inflammatory, anti-aging gene FOXO3 promotes longevity in humans: |
| ○ | Replicated in >20 independent studies | |
| ○ | Confers CVD protective benefit (p=0.001) | |
| ○ | Decreases inflammation (CRP, trend; TNF-α, p=0.018) |
In animal studies conducted by third parties and us, astaxanthin statistically significantly decreased activated the FOXO3 gene and extended lifespan:
| ● | FOXO3 mRNA levels increased in mice by 90% (p=0.024) | |
| ● | Lifespan extended by up to 30% via FOXO3 ortholog DAF16 in roundworms |
Our Products and Business Strategy
Our product platform consists of our development stage pharmaceutical candidates and our commercially available dietary supplement:
| ● | CDX-101, our lead pharmaceutical candidate, is in pre-clinical development for cardiovascular inflammation and dyslipidemia, with a target initial indication of severe hypertriglyceridemia. | |
| ● | CDX-301 is in pre-clinical development for macular degeneration, with a target initial indication of Stargardt disease. | |
| ● | ZanthoSyn® is a physician recommended astaxanthin dietary supplement for inflammatory health. |
| 38 |
Lead Pharmaceutical Candidate: CDX-101
Our lead pharmaceutical candidate, CDX-101, is a proprietary astaxanthin prodrug that cleaves following oral administration and delivers astaxanthin to the bloodstream. CDX-101 is being developed initially for cardiovascular inflammation and mixed dyslipidemia, with a target initial indication of severe hypertriglyceridemia.
We believe that the results from two major cardiovascular clinical trials—the 10,061 patient CANTOS study by Novartis in 2017 and the 8,179 patient REDUCE-IT study by Amarin in 2018—clearly demonstrated the clinical significance of reducing chronic inflammation, validating the cardiovascular inflammation hypothesis we have supported for more than a decade. We believe that astaxanthin’s unique mechanism of action—reduction of oxidative stress driven inflammation at the cellular and mitochondrial level without inhibiting normal function—results in an impact on key inflammatory drug targets and pathways, and importantly, an excellent safety profile that supports chronic administration. In addition to the safety advantages described in this prospectus, we believe that production of CDX-101, unlike Vascepa and other prescription fish oil drugs, will be highly scalable to address these large mass markets for chronic diseases driven by inflammation.
Clinical and non-clinical studies with astaxanthin have provided proof-of-concept for the treatment of cardiovascular risk factors including inflammation and triglycerides as described in this prospectus. In addition, interim results from our Cardiovascular Health Astaxanthin Supplement Evaluation (“CHASE”) clinical trial demonstrate beneficial changes in markers of cardiovascular health, including CRP, LDL cholesterol, total cholesterol, triglycerides, oxidized LDL, and blood pressure, and also underscore astaxanthin’s safety profile with no adverse safety signals observed. We believe these findings provide further mechanistic support for our pharmaceutical development program. We refer you to “CHASE Clinical Trial” on page 40 of this prospectus for additional information regarding the CHASE clinical trial.
We believe that an initial indication of severe hypertriglyceridemia provides an efficient clinical pathway to drug approval for CDX-101 and will be similar to the pathway as reported by Amarin for the development of Vascepa, its prescription fish oil. CDX-101 is currently in pre-clinical development, including the planning of IND enabling studies. We plan to use proceeds from this offering to complete IND enabling studies and to engage third party contract development and manufacturing organizations (CDMOs) to manufacture drug substance and drug product for such studies, with the goal of filing an IND approximately one year from the closing of this offering.
We have retained Paresh N. Soni, M.D., Ph.D., the former Senior Vice President and Head of Development at Amarin, to guide our clinical and regulatory strategy, interact with the FDA, and advise us on a full range of development issues. While at Amarin, Dr. Soni led the design of Amarin’s clinical trials, development strategy, and interaction with the FDA, including for Vascepa, which was approved for treatment of severe hypertriglyceridemia in 2012. Dr. Soni played a key role in the design and conduct of the MARINE, ANCHOR and REDUCE-IT clinical trials with Vascepa. In addition, Dr. Soni has held several senior R&D executive roles over the past 2 decades at Pfizer, Alexion, and Albireo. Dr. Soni is also a member of our Scientific Advisory Board.
In addition to Dr. Soni, our Scientific Advisory Board includes Deepak L. Bhatt, M.D., M.P.H. and R. Preston Mason, Ph.D.
Deepak L. Bhatt, M.D., M.P.H., is the Chairman of our Scientific Advisory Board. Dr. Bhatt is also the Chair of the REDUCE-IT clinical trial with Vascepa, Executive Director of Interventional Cardiovascular Programs at Harvard Medical School affiliated Brigham and Women’s Hospital, and Professor at Harvard Medical School. He is also the Editor of the peer-reviewed Journal of Invasive Cardiology and Editor-in-Chief of the Harvard Heart Letter for patients.
R. Preston Mason, Ph.D. is on the faculty of the Department of Medicine, Division of Cardiology at Harvard Medical School affiliated Brigham and Women’s Hospital. He has published more than 250 peer reviewed papers, including papers published in collaboration with Cardax, and is a recognized expert on the mechanism of action of astaxanthin and fish oils, particularly Vascepa.
CDX-101 vs. ZanthoSyn®
CDX-101 is a synthetic astaxanthin prodrug (new chemical entity) for pharmaceutical applications and ZanthoSyn® is a formulation of synthetic nature-identical astaxanthin for dietary supplement applications. While both deliver astaxanthin to the bloodstream, we believe the unique molecular structure of CDX-101 and its pharmaceutical pathway will provide substantial differentiation. In particular, we believe that:
| ● | CDX-101 will be approved by the FDA as a drug for one or more disease indications, whereas ZanthoSyn® is marketed as a dietary supplement for health applications; | |
| ● | CDX-101 will be prescribed by doctors and covered by health insurance, whereas ZanthoSyn® is sold through retail and e-commerce channels; | |
| ● | CDX-101 will be administered at a higher dose and in different oral dosage form; and | |
| ● | CDX-101 will have superior intellectual property protection. |
Pharmaceutical Candidate: CDX-301
Our zeaxanthin pharmaceutical candidate, CDX-301, has a mechanism of action and excellent safety profile similar to astaxanthin, however, it is being developed for macular degeneration because zeaxanthin accumulates in the human eye through uptake by a unique retinal receptor, providing protection against blue light, oxidative damage, and related inflammation that occurs in macular degeneration. Pre-clinical and clinical studies with zeaxanthin have demonstrated proof-of-concept for the treatment of macular disorders. We believe that an initial indication of Stargardt disease, a juvenile form of macular degeneration, provides an efficient clinical pathway to drug approval for CDX-301. On November 30, 2018, we submitted a request for orphan drug designation to the FDA for zeaxanthin as a treatment of Stargardt disease, and we are currently in communications with the FDA regarding this matter. Additional financing beyond that contemplated in this offering will be needed to fund IND enabling studies and clinical development of CDX-301.
Dietary Supplement: ZanthoSyn®
ZanthoSyn® is our commercially available physician recommended astaxanthin dietary supplement. Astaxanthin is a naturally occurring molecule with safe anti-inflammatory activity that supports cardiovascular health, metabolic health, liver health, joint health, and longevity. The form of astaxanthin utilized in ZanthoSyn® has demonstrated an excellent safety profile in peer-reviewed published studies and is GRAS according to FDA regulations.
We sell ZanthoSyn® primarily through wholesale and e-commerce channels. We launched our e-commerce channel in 2016 and began selling to GNC stores in 2017. ZanthoSyn® is currently available at GNC corporate stores nationwide in the United States.
ZanthoSyn® is the top selling product at GNC stores in Hawaii and the top selling product in the anti-oxidant category at GNC stores nationwide.
| 39 |
We market ZanthoSyn® primarily through a multi-pronged approach:
| ● | Physician outreach and education, where ZanthoSyn® is positioned as the first safe, physician friendly, anti-inflammatory dietary supplement for health and longevity, with retail locations and e-commerce serving as convenient and credible distribution channels for physicians recommending ZanthoSyn® | |
| ● | Retail store outreach, education, and in-store sales support, building on the ability to utilize ZanthoSyn® as a foundation of health and wellness regimens | |
| ● | E-commerce platforms |
We believe ZanthoSyn® is physician friendly for several reasons:
| ● | ZanthoSyn® delivers the safety, purity, manufacturing rigor, bioavailability, and scientific support that provides physicians comfort in the quality and utility of the product, which is often not present in other dietary supplements. | |
| ● | ZanthoSyn® is well-accepted at medical conferences where crowds of physicians and other healthcare professionals stand in line to receive ZanthoSyn® samples and product information after attending educational seminars. |
Our sales and marketing program was initially launched in Hawaii, where we believe that robust physician outreach and education coupled with GNC retail store outreach, education, and in-store sales support increased consumer awareness and catalyzed strong sales growth. We also launched this program in major markets on the West Coast and East Coast in the U.S. beginning in 2017. To support these efforts, we have hired additional sales and marketing personnel. We are currently evaluating our strategy related to further expansion.
We sell ZanthoSyn® to GNC under a purchasing agreement. The exclusivity provision under such agreement related to distribution of ZanthoSyn® by GNC in the “brick and mortar” retail channel in the United States expired on October 16, 2019. GNC remains our only distributor of ZanthoSyn® in such channel, but we may expand retail distribution to mass market retailers, other specialty nutrition stores, pharmacies, and other retailers. We also plan to increase our sales and marketing efforts through e-commerce.
To date, our sales and marketing efforts of ZanthoSyn® have primarily been through GNC retail store outreach, education, and in-store sales support together with physician outreach and education. We plan to increase our sales and marketing efforts through e-commerce by capitalizing on one of the most important lessons learned from our sales and marketing program: “Conversations Create Customers.” Whether at GNC stores, directly with Cardax personnel, or at conferences with healthcare professionals, thousands of ZanthoSyn® customers have been created by understanding and experiencing the benefits of ZanthoSyn® firsthand. Cardax plans to implement strategies that it believes may create a similar customer experience more broadly, with fulfillment online, where margins may be greater than retail stores.
CHASE Clinical Trial
In September 2018, we initiated a human clinical trial entitled, Cardiovascular Health Astaxanthin Supplement Evaluation (“CHASE”), targeting cardiovascular inflammatory health. The randomized, double-blind, placebo-controlled clinical trial is evaluating the effect of low-dose and high-dose ZanthoSyn® on cardiovascular health as measured by CRP levels over 12 weeks in up to 120 subjects with documented cardiovascular risk factors. The study also includes an optional open label extension through 48 weeks.
Interim results from an initial cohort of subjects were announced on September 23, 2019. The interim results were based on data from 40 subjects administered high-dose ZanthoSyn® (96 mg/day astaxanthin – 48 mg twice a day), low-dose ZanthoSyn® (24 mg/day astaxanthin – 12 mg twice a day), or placebo.
Highlights from the interim review shown below are median percentage changes from baseline to week 12 unless otherwise stated. While the interim review was not powered for statistical significance, p-values less than 0.05 compared to placebo are provided. The p-values reported below (*p<0.05, **p<0.01) are nominal p-values from non-parametric comparisons of the median between each group and placebo and no adjustments for multiple comparisons were made.
| Interim Results | High Dose | Low Dose | Placebo | |||||||||
| CRP | -28% | -32% | -5% | |||||||||
| LDL-C | -12% | ** | -7% | +5% | ||||||||
| Total cholesterol | -8% | * | -5% | +4% | ||||||||
| Triglycerides | -16% | -13% | +6% | |||||||||
| Oxidized LDL | -10% | * | +3% | +4% | ||||||||
| Blood pressure | -5% | * | -4% | * | +6% | |||||||
| Median astaxanthin blood levels at 12 weeks | 2,184 ng/mL | 790 ng/mL | <10 ng/mL | |||||||||
We believe these findings provide:
| ● | Further mechanistic support for our astaxanthin pharmaceutical development program | |
| ● | Basis for additional patent filings | |
| ● | Support for the cardiovascular health benefits of ZanthoSyn® |
The interim results also underscore astaxanthin’s safety profile with no adverse safety signals observed. The CHASE Data Safety Review Board, which is comprised of a majority of independent clinical trial professionals, recommended that the clinical trial continue enrollment.
The FDA does not require human clinical trials for dietary supplements, but we believe that positive results from the CHASE trial may help promote scientific and consumer awareness of astaxanthin’s health and longevity applications and serve as further mechanistic support for our pharmaceutical development program.
We refer to you the “Risk Factors” section of this prospectus for a summary of certain risks related to clinical trial results.
REPZ Clinical Study
We are also exploring the effect of ZanthoSyn® on recovery, endurance, and performance in a clinical study (the Recovery, Endurance, and Performance with ZanthoSyn® or “REPZ” study) with 40 subjects by measuring sprint times and heart rates in connection with high intensity interval training on stationary air bikes. The results of the REPZ study, if successful, may be used to support ZanthoSyn® marketing efforts for sports and fitness applications. The REPZ study was recently completed, and final data analysis is underway.
| 40 |
Benefits of Synthetic Astaxanthin vs. Natural Astaxanthin
Dietary supplements containing astaxanthin typically derive astaxanthin from microalgae, krill, or other natural sources, whereas ZanthoSyn® astaxanthin is made through total synthesis. While multiple studies demonstrate that astaxanthin from either natural or synthetic sources is efficacious and both are Generally Recognized as Safe according to FDA regulations, we believe synthetic astaxanthin offers significant advantages compared to astaxanthin from microalgae, krill, or other natural sources:
| ● | Synthetic astaxanthin can be formulated for superior bioavailability. In a human crossover study comparing ZanthoSyn® to a leading microalgal astaxanthin dietary supplement, the astaxanthin blood levels following administration of ZanthoSyn® were nearly three times higher than the microalgal astaxanthin product at the same dose: |
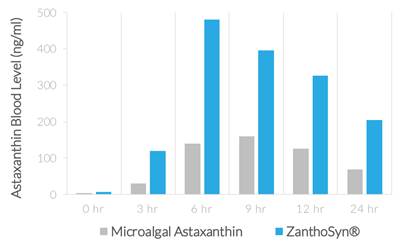
| ● | AUC (area under curve, astaxanthin blood levels) = 2.85-fold greater (p=0.013) | |
| ● | Cmax (maximum concentration, astaxanthin blood levels) = 3.0-fold greater (p=0.013) | |
| ● | Coefficient of variation (variation between subjects of astaxanthin blood levels) |
| ○ | ZanthoSyn® = 27% | |
| ○ | Microalgal astaxanthin = 62% |
| ● | Tmax (time of maximum concentration) = 6 hours | |
| ● | No adverse events observed |
The superior bioavailability described in this prospectus means that three times more astaxanthin from ZanthoSyn® is absorbed into the body from each dose, which provides a superior value proposition compared to other astaxanthin dietary supplements.
| ● | Synthetic astaxanthin has been extensively tested in a wide range of toxicity studies, including acute, subacute, sub-chronic, and chronic toxicity studies, carcinogenicity studies, genotoxicity/mutagenicity studies, and developmental and reproductive toxicity studies; whereas to our knowledge microalgal or other sources of astaxanthin have not undergone the same amount of safety testing in such toxicity studies. | |
| ● | Synthetic astaxanthin is manufactured with superior purity and precision, whereas astaxanthin extracted from microalgae and krill oil is obtained in a complex mixture, which may include many unknown marine byproducts. | |
| ● | Synthetic manufacture of astaxanthin is scalable, whereas we believe the ability to readily scale the production and extraction of astaxanthin from microalgae or other sources will be limited as demand for astaxanthin grows. | |
| ● | Synthetic manufacture of astaxanthin emits fewer greenhouse gases and consumes less energy, raw material, and land than traditional microalgal astaxanthin production. |
Intellectual Property
We have obtained and are continuing to seek patent protection for compositions of matter, pharmaceutical compositions, and pharmaceutical uses, in certain disease areas, of our various carotenoid analogs and derivatives. Such carotenoids include astaxanthin, zeaxanthin, lutein, and/or lycophyll, and esters and other analogs and derivatives of these compounds. More specifically, we seek to protect: (i) the composition of matter of novel carotenoid analogs and derivatives, (ii) pharmaceutical compositions comprising synthetic or natural preparations of novel or natural occurring carotenoid analogs and derivatives, and (iii) the pharmaceutical use of synthetic preparations of novel or naturally occurring carotenoid analogs and derivatives in specific disease areas, including, but not limited to, the treatment of inflammation and related tissue damage, liver disease, and reperfusion injury, as well as the pharmaceutical use of synthetic or natural preparations of novel or natural occurring carotenoid analogs and derivatives for the reduction of platelet aggregation. We intend to enforce and defend our intellectual property rights consistent with our strategic business objectives.
We have 29 issued patents and two pending patents related to the composition of matter, pharmaceutical compositions, and pharmaceutical uses of our drugs candidates as well as many other related molecules that will expire between 2023 and 2028, subject to patent term extensions. We also have filed additional patents to extend patent coverage in the U.S. and worldwide to 2039-2040, with such applications including coverage related to certain cardiovascular uses on the basis of the CHASE clinical trial results as well as coverage related to the composition of matter of CDX-101.
| 41 |
The Company’s patents are summarized in the table below.
| United States | Foreign | Expiration | ||||||||||
| Issued Patents | 14 | 15 | 2023-2028 | |||||||||
| Pending Patents | 0 | 1 | 2023-2028 | |||||||||
| Pending Patents | 2 | 1 | 2039-2040 | |||||||||
Research and Development
Our research and development program is presently comprised of employees, consultants, including regulatory, scientific, and medical professionals, and third-party collaborators or contract organizations, including academic institutions, contract research organizations, and contract development and manufacturing organizations. Contract organizations provide us with access to significant research and development resources and infrastructure. We anticipate that our research and development will be primarily conducted by contract organizations with direction and oversight by our in-house research and development personnel.
In addition to conducting or overseeing research and development activities, our research and development personnel analyze and interpret other research on astaxanthin, as well as related compounds, competing products, applications, and industry trends. In the United States National Library of Medicine’s online repository, PubMed.gov, there are more than 2,000 peer-reviewed journal articles that reference astaxanthin in the title or abstract, over 500 of which were published in the last three years, with the vast majority published by organizations and researchers that are not affiliated with us. This type of “open-source” research has served to significantly advance the understanding of astaxanthin and related carotenoids, and has also presented our research and development personnel with the critical task of keeping up-to-date on all of the latest research and interpreting and integrating the findings with our research and that of others in order to serve as leading experts on the mechanism of action and biological applications of astaxanthin and related carotenoids.
Our research and development expenditures totaled $269,077 and $97,479 for the years ended December 31, 2018 and 2017, respectively, and we incurred $250,141 of research and development expenditures during the nine-month period ending September 30, 2019. These expenditures primarily reflect the cost of product development activities, including clinical trials. The compensation of our research and development personnel are included as a component of salaries and wages in the consolidated statements of operations. Our research and development expenditures for the year ended December 31, 2017 were reclassified to conform to the presentation of expenditures for the year ended December 31, 2018, in accordance with Note 2 to such consolidated financial statements.
Manufacturing
We utilize contract manufacturers and/or other third-party suppliers for the production of our products. The raw materials and supplies required for the production of our products may be available, in some instances from one supplier, and in other instances, from multiple suppliers. In those cases where raw materials are only available through one supplier, such supplier may be either a sole source (the only recognized supply source available to us) or a single source (the only approved supply source for us among other sources). We, our contract manufacturers, and/or other third-party suppliers will adopt appropriate policies to attempt, to the extent feasible, to minimize our raw material supply risks, including maintenance of greater levels of raw materials inventory and implementation of multiple raw materials sourcing strategies, especially for critical raw materials. Although to date we have not experienced any significant delays in obtaining any raw materials from suppliers, we cannot provide assurance that we, our contract manufacturers, and/or other third-party suppliers will not face shortages from one or more suppliers in the future.
Competition
The industries in which we intend to compete are subject to intense competition. The primary competition for our pharmaceutical candidates are the numerous pharmaceutical and biotechnology companies developing or marketing anti-inflammatories and other drugs or therapeutics targeting chronic diseases driven by inflammation, including cardiovascular disease, metabolic disease, liver disease, arthritis, aging, and macular degeneration. Certain competitors for our pharmaceutical candidates include, but are not limited to, AbbVie, Acasti Pharma, Acucela, Alkeus Pharmaceuticals, Amarin, Amgen, Astellas, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Gilead, GlaxoSmithKline, Johnson & Johnson, Matinas Biopharma, Merck, MT Pharma, Nestle/Pamlab, Novartis, Pfizer, Reata Pharmaceuticals, Regeneron Pharmaceuticals, Roche/Genentech, Sanofi-Aventis, Servier, and Takeda. The primary competition for our dietary supplements are the many companies developing or marketing astaxanthin dietary supplements and other supplements targeting inflammatory health, cardiovascular health, metabolic health, liver health, joint health, and longevity. We believe that our ability to compete will be based primarily on scientific superiority, availability of patent protection, protection of other intellectual property rights, access to adequate capital, ability to develop, acquire, and market products successfully, ability to obtain governmental approvals, and ability to serve the particular needs of customers.
Our success will depend in large part on our ability to obtain and maintain international and domestic patents, other intellectual property, and other legal protections for the proprietary technology that we consider important to our business. We intend to continue to seek appropriate patent protection for our products where applicable by filing patent applications in the United States and other selected countries. We intend for these patent applications to cover, where applicable, claims for composition of matter, uses, manufacturing processes, and formulations. Our success will also depend on our ability, and the ability of our current and/or future strategic partners to maintain intellectual property rights related to proprietary production methods for products that we, or our partners, intend to market.
Government Regulation
Most aspects of our business are subject to some degree of government regulation. For some of our products, government regulation is significant and, in general, there appears to be a trend toward more stringent regulation throughout the world, as well as global harmonization of various regulatory requirements. We expect to devote significant time, effort and expense to address the extensive government and regulatory requirements applicable to our business. We believe that we are no more or less adversely affected by existing government regulations than our competitors.
| 42 |
FDA Regulation
Biopharmaceutical companies must comply with comprehensive regulation by the FDA and other regulatory agencies in the United States and comparable authorities in other countries. While the FDA does not require human clinical trials for dietary supplements, we have conducted and may continue to conduct clinical trials with our dietary supplements to promote scientific and consumer awareness. We may also conduct Phase I, Phase II, and/or Phase III clinical trials with our pharmaceutical candidates.
We must obtain regulatory approvals by the FDA and similar health authorities in foreign countries to the extent applicable prior to human clinical testing and marketing of any pharmaceutical and for post-approval clinical studies for additional indications of approved drugs. We anticipate that any pharmaceutical candidate will be subject to rigorous preclinical and clinical testing and pre-market approval procedures by the FDA and similar health authorities in foreign countries to the extent applicable. The extent to which our products are regulated by the FDA will depend upon the types of products we ultimately develop. We are currently evaluating and pursuing various developmental strategies and cannot predict, during this stage of our development, the scope of FDA or other agency regulation to which we or our products will be subject. Various federal statutes and regulations also govern or influence the preclinical and clinical testing, record-keeping, approval, labeling, manufacture, quality, shipping, distribution, storage, marketing and promotion, export, and reimbursement of pharmaceuticals.
The steps ordinarily required before a drug product may be marketed in the United States include:
| ● | preclinical studies; | |
| ● | submission to the FDA of an IND application, which must become effective before human clinical trials may commence; | |
| ● | adequate and well-controlled human clinical trials to establish the safety and efficacy of the pharmaceutical candidate in the desired indication for use; | |
| ● | submission to the FDA of a NDA, together with payment of a substantial user fee; and | |
| ● | FDA approval of the NDA, including inspection and approval of the product manufacturing facility and select sites at which human clinical trials were conducted. |
Preclinical studies typically involve laboratory evaluation of pharmaceutical candidate chemistry, formulation, and stability, as well as animal studies to assess the potential safety and efficacy of the pharmaceutical candidate. The results of preclinical studies are submitted to the FDA as part of an IND and are reviewed by the FDA before the commencement of clinical trials. Unless the FDA objects to an IND, the IND will become effective 30 days following its receipt by the FDA. Submission of an IND may not result in FDA clearance to commence clinical trials, and the FDA’s failure to object to an IND does not guarantee FDA approval of a marketing application.
Clinical trials involve the administration of the test agent to humans under the supervision of a qualified principal investigator. In the United States, clinical trials must be conducted in accordance with Good Clinical Practices. In addition, each clinical trial must be approved and conducted under the auspices of an institutional review board and with the subject’s informed consent. We would be subject to similar regulatory considerations if we conduct clinical trials outside the United States.
The goal of Phase I clinical trials is to establish initial data about safety and tolerability of the pharmaceutical candidate in humans. The investigators seek to evaluate the effects of various dosages and to establish an optimal dosage level and schedule.
The goal of Phase II clinical trials is to provide evidence about the desired therapeutic efficacy of the pharmaceutical candidate in limited studies with small numbers of carefully selected subjects. Investigators also gather additional safety data.
Phase III clinical trials consist of expanded, large-scale, multi-center studies in the target patient population. This phase further tests the product’s effectiveness, monitors side effects, and, in some cases, compares the product’s effects to a standard treatment, if one is already available. Phase III trials are designed to more rigorously test the efficacy of a pharmaceutical candidate and are normally randomized, double-blinded, and placebo-controlled. Phase III trials are typically monitored by an independent data monitoring committee, or DMC, which periodically reviews data as a trial progresses. A DMC may recommend that a trial be stopped before completion for a number of reasons including safety concerns, patient benefit, or futility.
Data obtained from this development program are submitted as part of an NDA to the FDA and possibly to corresponding agencies in other countries for review. The NDA requires agency approval prior to marketing in the relevant country. Extensive regulations define the form, content and methods of gathering, compiling and analyzing the pharmaceutical candidate’s safety and efficacy data.
The process of obtaining regulatory approval can be costly, time consuming and subject to unanticipated delays. Regulatory agencies may refuse to approve an application if they believe that applicable regulatory criteria are not satisfied and may also require additional testing for safety and efficacy and/or post-marketing surveillance or other ongoing requirements for post-marketing studies. In some instances, regulatory approval may be granted with the condition that confirmatory Phase IV clinical trials are carried out, and if these trials do not confirm the results of previous studies, regulatory approval for marketing may be withdrawn. Moreover, each regulatory approval of a product is limited to specific indications. The FDA or other regulatory authorities may approve only limited label information for the product. The label information describes the indications and methods of use for which the product is authorized, may include Risk Evaluation and Mitigation Strategies and, if overly restrictive, may limit a sponsor’s ability to successfully market the product. Regulatory agencies routinely revise or issue new regulations, which can affect and delay regulatory approval of pharmaceuticals.
Furthermore, pharmaceutical manufacturing processes must conform to current Good Manufacturing Practices, or cGMPs. Manufacturers, including a drug sponsor’s third-party contract manufacturers, must expend time, money and effort in the areas of production, quality control and quality assurance, including compliance with stringent record-keeping requirements. Manufacturing establishments are subject to periodic inspections by the FDA or other health authorities, in order to assess, among other things, compliance with cGMP. Before approval of the initiation of commercial manufacturing processes, the FDA will usually perform a preapproval inspection of the facility to determine its compliance with cGMP and other rules and regulations. In addition, foreign manufacturers must also comply with cGMPs in order to supply products for use in the United States, and are subject to periodic inspection by the FDA or by regulatory authorities in certain countries under reciprocal agreements with the FDA. Manufacturing processes and facilities for pharmaceuticals are highly regulated. Regulatory authorities may choose not to certify or may impose restrictions, or even shut down existing manufacturing facilities that they determine are non-compliant.
| 43 |
FDA GRAS Determination
“GRAS” is an acronym for the phrase “generally recognized as safe,” which the FDA utilizes to describe those substances that, in the generally recognized opinion of the scientific community, will not be harmful to consumers, provided the substance is used as intended. According to applicable FDA regulations, any substance that is intentionally added to food is a food additive, which is subject to premarket review and approval by FDA, unless the substance is generally recognized, among qualified experts, as having been adequately shown to be safe under the conditions of its intended use. Under sections 201(s) and 409 of the Federal Food, Drug, and Cosmetic Act (the “FD&C Act”), and FDA’s implementing regulations in 21 CFR 170.3 and 21 CFR 170.30, the use of a food substance may be GRAS either through scientific procedures or, for a substance used in food before 1958, through experience based on common use in food. General recognition of safety through scientific procedures requires the same quantity and quality of scientific evidence as is required to obtain approval of the substance as a food additive and ordinarily is based upon published studies, which may be corroborated by unpublished studies and other data and information. General recognition of safety through experience based on common use in foods requires a substantial history of consumption for food use by a significant number of consumers.
Manufacturers of GRAS substances may provide the FDA with a notification of GRAS determination, which includes a description of the substance, the applicable conditions of use, and an explanation of how the substance was determined to be safe. Upon review of such a notification, the FDA may respond with a “no questions” position, whereby the manufacturer’s determination that a product is GRAS for its intended purposes is affirmed. Alternatively, manufacturers may elect to “self-affirm” a given substance as GRAS without FDA notification but should retain all applicable safety data used for GRAS determination in the case of FDA inquiry.
Synthetic copies of naturally-occurring dietary ingredients or related components do not qualify as dietary ingredients under the FD&C Act, but substances that have been affirmed by the FDA as GRAS, self-affirmed as GRAS, or approved as direct food additives in the U.S. may be marketed as dietary ingredients, subject to FDA regulations for dietary ingredients.
FDA NDI Notification
The Dietary Supplement Health and Education Act of 1994 (the “DSHEA”) (Pub. L. 103-417) was signed into law on October 25, 1994 and amended the FD&C Act by adding: (i) section 201(ff) (21 U.S.C. 321(ff)), which defines the term “dietary supplement”, and (ii) section 413 (21 U.S.C. 350b), which defines the term “new dietary ingredient” (“NDI”) and requires the manufacturer or distributor of an NDI, or of the dietary supplement that contains the NDI, to submit a premarket notification to FDA at least 75 days before introducing/delivering the supplement into interstate commerce, unless the NDI and any other dietary ingredients in the dietary supplement have been present in the food supply without chemical alteration (21 U.S.C. 350b(a)(1)). The NDI notification must contain applicable information, including history of use and citations to published articles, from which the manufacturer or distributor of the NDI or dietary supplement has concluded that the dietary supplement containing the NDI will be reasonably expected to be safe under the conditions of its intended use. NDI notifications are not required for the marketing of approved food additives or GRAS substances as NDIs unless the dietary ingredient has been chemically altered.
FDA Orphan Drug Designation
The Orphan Drug Act was signed into law on January 4, 1983. The Congressional findings for the Orphan Drug Act were as follows: (i) there are many rare diseases and conditions that affect such small numbers of individuals residing in the United States; (ii) adequate drugs for many rare diseases and conditions have not been developed; (iii) drugs for rare diseases and conditions are commonly referred to as “orphan drugs”; (iv) because so few individuals are affected by any one rare disease or condition, a pharmaceutical company that develops an orphan drug may reasonably expect the drug to generate relatively small sales in comparison to the cost of developing the drug and consequently to incur a financial loss; (v) there is reason to believe that some promising orphan drugs will not be developed unless changes are made in the applicable Federal laws to reduce the costs of developing such drugs and to provide financial incentives to develop such drugs; and (vi) it is in the public interest to provide such changes and incentives for the development of orphan drugs.
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition that (i) affects less than 200,000 persons in the United States, or (ii) affects more than 200,000 in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug for such disease or condition will be recovered from sales in the United States of such drug. Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the identity of the drug and its potential orphan use are disclosed publicly by the FDA.
In the United States, orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages, and NDA user-fee waivers. In addition, if a drug receives the first FDA approval for the indication for which it has orphan designation, the drug is entitled to orphan drug exclusivity, which means the FDA may not approve any other application, including a full NDA, to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority over the drug with orphan exclusivity or where the manufacturer with orphan exclusivity is unable to assure sufficient quantities of the approved orphan-designated drug. Competitors, however, may receive approval of different drugs for the indication that the orphan drug has exclusivity or obtain approval for the same drug but for a different indication for which the orphan drug has exclusivity. Orphan drug exclusivity also could block the approval of one of our drugs for seven years if a competitor obtains approval of the same drug as defined by the FDA or if our drug is determined to be contained within the competitor’s drug for the same indication or disease. If a drug designated as an orphan drug receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan drug exclusivity. In addition, exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the drug to meet the needs of patients with the rare disease or condition. There can be no assurance that any request for orphan drug designation will be granted by the FDA.
| 44 |
Other Regulations
Pharmaceutical companies are subject to various federal and state laws pertaining to healthcare “fraud and abuse,” including anti-kickback and false claims laws. The Anti-Kickback Statute is a federal criminal statute that makes it illegal for any person, including a prescription drug manufacturer, or a party acting on its behalf, to knowingly and willfully solicit, offer, receive or pay any remuneration, directly or indirectly, in exchange for, or to induce, the referral of business, including the purchase, order or prescription of a particular drug, for which payment may be made under federal healthcare programs such as Medicare and Medicaid. Some of the state prohibitions apply to referral of patients for healthcare services reimbursed by any source, not only the Medicare and Medicaid programs.
In the course of practicing medicine, physicians may legally prescribe FDA approved drugs for an indication that has not been approved by the FDA and which, therefore, is not described in the product’s approved labeling, so-called “off-label use.” The FDA does not ordinarily regulate the behavior of physicians in their choice of treatments. The FDA and other governmental agencies do, however, restrict communications on the subject of off-label use by a manufacturer or those acting on behalf of a manufacturer. Companies may not promote FDA-approved drugs for off-label uses. The FDA and other governmental agencies do permit a manufacturer (and those acting on its behalf) to engage in some limited, non-misleading, non-promotional exchanges of scientific information regarding unapproved indications. The United States False Claims Act prohibits, among other things, anyone from knowingly and willfully presenting, or causing to be presented for payment to third-party payers (including Medicare and Medicaid) claims for reimbursed drugs or services that are false or fraudulent, claims for items or services not provided as claimed or claims for medically unnecessary items or services. Violations of fraud and abuse laws may be punishable by criminal and/or civil sanctions, including imprisonment, fines and civil monetary penalties, as well as possible exclusion from federal health care programs (including Medicare and Medicaid). In addition, under this and other applicable laws, such as the Food, Drug and Cosmetic Act, there is an ability for private individuals to bring similar actions. Further, there is an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information. Many of these laws contain ambiguities as to what is required to comply with the law.
We are subject to various laws and regulations regarding laboratory practices and the experimental use of animals in connection with our research. In each of these areas, as above, the FDA and other regulatory authorities have broad regulatory and enforcement powers, including the ability to suspend or delay issuance of approvals, seize or recall products, withdraw approvals, enjoin violations and institute criminal prosecution, any one or more of which could have a material adverse effect upon our business, financial condition, and results of operations.
We must comply with regulations under the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act and other federal, state and local regulations. We are subject to federal, state and local laws and regulations governing the use, generation, manufacture, storage, air emission, effluent discharge, handling and disposal of certain hazardous or potentially hazardous materials. We may be required to incur significant costs to comply with environmental and health and safety regulations in the future. Our research and development involves the controlled use of hazardous materials, including, but not limited to, certain hazardous chemicals.
Our activities are also potentially subject to federal and state consumer protection and unfair competition laws. We are also subject to the United States Foreign Corrupt Practices Act, or the FCPA, which prohibits companies and individuals from engaging in specified activities to obtain or retain business or to influence a person working in an official capacity. Under the FCPA, it is illegal to pay, offer to pay, or authorize the payment of anything of value to any foreign government official, governmental staff members, political party or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity. In addition, federal and state laws protect the confidentiality of certain health information, in particular, individually identifiable information, and restrict the use and disclosure of that information. At the federal level, the Department of Health and Human Services promulgated health information privacy and security rules under the Health Insurance Portability and Accountability Act of 1996. In addition, many state laws apply to the use and disclosure of health information.
Customers
We sell ZanthoSyn® primarily through wholesale and e-commerce channels. We launched our e-commerce channel in 2016, and we began selling to GNC stores in 2017. ZanthoSyn® is currently available at over 3,000 GNC corporate stores in the United States.
During the years ended December 31, 2018 and 2017, sales to GNC accounted for more than 90% and more than 75% of our revenues, respectively. During the nine months ended September 30, 2019, sales to GNC account for more than 80% of our revenues. No other customer accounted for 10% or more of our revenues during these years.
We sell ZanthoSyn® to GNC under a purchasing agreement. The exclusivity provision under such agreement related to distribution of ZanthoSyn® by GNC in the “brick and mortar” retail channel in the United States expired on October 16, 2019. GNC remains our only distributor of ZanthoSyn® in such channel, but we may expand retail distribution to mass market retailers, other specialty nutrition stores, pharmacies, and other retailers. We also plan to increase our sales and marketing efforts through e-commerce.
Employees
As of the date of this prospectus, we have 11 full-time employees and one part-time employee. None of our employees are subject to a collective bargaining agreement. We believe the relations with our employees are satisfactory.
Properties
We maintain a facility of approximately 738 square feet at 2800 Woodlawn Drive, Honolulu, Hawaii, which is leased on a month-to-month basis. We believe that our facility is adequate for our current purposes.
Legal Proceedings
From time to time, we may become involved in various lawsuits and legal proceedings that arise in the ordinary course of business. However, litigation is subject to inherent uncertainties and an adverse result in these or other matters may arise from time to time that may harm our business. We are currently not aware of any such legal proceedings or claims that we believe will have a material adverse effect on our business, financial condition or operating results.
| 45 |
Directors, Executive Officers, and Corporate Governance
Set forth below is a list of the names, ages and positions of our directors and executive officers.
| Name | Age | Position(s) | ||
| George W. Bickerstaff, III | 64 | Chairman of the Board of Directors | ||
| David G. Watumull | 69 | President, Chief Executive Officer, and Director | ||
| Terence A. Kelly, Ph.D. | 58 | Director | ||
| Michele Galen | 63 | Director | ||
| Makarand Jawadekar, Ph.D. | 68 | Director | ||
| Elona Kogan | 50 | Director | ||
| David M. Watumull | 37 | Chief Operating Officer | ||
| John B. Russell | 47 | Chief Financial Officer and Treasurer | ||
| Richard M. Morris | 59 | Secretary |
Biographies of Directors and Executive Officers
George W. Bickerstaff, III has served as a Director since June 16, 2014. Mr. Bickerstaff is currently a partner and the managing director of M.M. Dillon & Co., a healthcare and technology investment bank that he co-founded. Previously, he served as Chief Financial Officer of Novartis Pharma AG from 2000 to 2005, held senior financial positions at IMS Health from 1989 to 1997 and held financial positions with Dun & Bradstreet and General Electric. Mr. Bickerstaff currently serves as a member of the boards of directors of the following public companies: Axovant Sciences Ltd., CareDx, Inc. and Innoviva, Inc. He also previously served on the board of directors of ARIAD Pharmaceuticals, Inc. and Inovio Pharmaceuticals, Inc. Mr. Bickerstaff received his B.S. in Engineering and his B.A. in Business Administration from Rutgers University. We believe that Mr. Bickerstaff’s experience in the pharmaceutical and biopharmaceutical industries and board leadership qualify him to serve on our Board of Directors.
David G. Watumull has served as our Chief Executive Officer, President, and Director since February 7, 2014. Mr. Watumull has served as the Chief Executive Officer, President, and Director of Cardax Pharma, Inc. since its inception in May 2013. Mr. Watumull also served as the Chief Executive Officer, President, and Director of Cardax Pharmaceuticals, Inc. from its inception in March 2006 until it merged with us in December 2015. Mr. Watumull is a co-founder of Cardax Pharmaceuticals, Inc. and has over 20 years of experience as a biotechnology industry executive. From 2001 to 2006, Mr. Watumull served as President, Chief Executive Officer, and Director of Hawaii Biotech, Inc. Mr. Watumull was Executive Vice President of Aquasearch, Inc., a public astaxanthin consumer health company, from 1998 to 2000. From 1997 to 1998 he headed his own biotech research firm, Watumull & Co. From 1994 to 1997 he was a biotech research analyst, money manager, and investment banker at First Honolulu Securities. From 1992 to 1994 he led his own money management firm, Biovest, Inc. Prior to that, from 1982 to 1992, Mr. Watumull worked at Paine Webber in various capacities, including as a biotech money manager and investment executive. Mr. Watumull studied mathematics at Claremont Men’s College (now Claremont McKenna College). Mr. Watumull’s extensive background in the biotechnology industry, his operational acumen, and his position of leadership since the founding of our business uniquely qualifies him to serve as a member of our Board of Directors.
Terence A. Kelly, Ph.D. has served as a Director since June 16, 2014. Dr. Kelly has over 25 years of experience as a scientist and executive in the pharmaceutical industry starting as a medicinal chemist in 1990. From 2011 to 2017, Dr. Kelly was the President and Chief Executive Officer of CoMentis, Inc. and currently acts as a consultant to the biotech industry through his company, Kelly Pharma Research Consulting, LLC. From 1990 to 2009, Dr. Kelly served in various scientific and executive positions at Boehringer Ingelheim, where after a successful early scientific career, he was promoted to Vice President of its U.S.-based medicinal chemistry department, which included 145 scientists in the high throughput screening, computational chemistry, structural biology, combinatorial chemistry and medicinal chemistry groups. Dr. Kelly holds a B.S. degree in Chemistry from Rensselaer Polytechnic Institute (1982) and a Ph.D. degree in Chemistry from the University of Texas at Austin (1988). He completed postdoctoral work in natural products synthesis at Yale University (1988-1990) and holds an M.B.A. from New York University, Stern School of Business (1998). Dr. Kelly is the co-author of over 25 scientific publications and served on the College of Natural Sciences Advisory Council for the University of Texas. Dr. Kelly’s scientific training and his track record of delivering high quality compounds into advanced clinical studies provide valuable skills and knowledge to our Board of Directors.
Michele Galen has served as a Director since January 4, 2017. Ms. Galen serves as a strategic advisor and board member across pharmaceuticals, biotechnology, health start-ups and global health, drawing on her broad experience in global business, communications, law and journalism. From June 2016 to present, Ms. Galen has led an independent consultancy, Michele Galen LLC. From April 2015 to June 2016, Ms. Galen served as Global Head, Communications and Public Affairs, for Shire plc, a biotechnology company, where she served as the lead communications and public affairs advisor on the successful $32 billion acquisition and integration of Baxalta. From February 2015 to March 2015, Ms. Galen led an independent consultancy, Michele Galen LLC. From May 2014 to January 2015, Ms. Galen served as a senior advisor to Novartis AG. From February 2012 to May 2014, Ms. Galen led Global Communications for Novartis AG, based in Basel, Switzerland. From February 2010 to February 2012, Ms. Galen served as Vice President and Global Head of Communications & Patient Advocacy for Novartis Pharma AG. From October 2003 to February 2010, Ms. Galen served as Vice President and Global Head, Oncology Affairs for Novartis Pharma AG. From February 2001 to October 2003, Ms. Galen served as Vice President, Corporate Communications for Novartis Pharmaceuticals Corporation. Earlier in her career, Ms. Galen was a Managing Director in the global public relations firm Burson-Marsteller. There, she co-founded the Organizational Change Communications practice. She is an award-winning journalist, and worked as Legal Editor and Social Issues Editor at Business Week magazine. Ms. Galen is a member of the New York State Bar and practiced law at Stroock, Stroock & Lavan LLP, and Skadden, Arps, Slate, Meagher & Flom LLP. Ms. Galen currently serves on the boards of Symphony Space and IYNAUS US. She formerly served on the advisory board of MK&A, Global Oncology, Stupid Cancer, and the Global Health Council. Ms. Galen received a B.A. from George Washington University, M.S. from the Columbia University Graduate School of Journalism, and J.D. from New York University School of Law. She also received a certification in executive coaching from Columbia University. Ms. Galen’s broad pharmaceutical, biotechnology, and healthcare background provide valuable skills and knowledge to our Board of Directors.
| 46 |
Makarand Jawadekar, Ph.D. has served as a Director since June 1, 2018. Dr. Jawadekar is a pharmaceutical executive with over 35 years of experience focused on research and development. From October 2017 to present, Dr. Jawadekar has served as Director and Chief Science Officer of Preveceutical Medical Inc., a Canadian pharmaceutical research and development company. Dr. Jawadekar also serves as a strategic advisor to pharmaceutical and biotechnology companies through his independent consultancy, Melinda Consulting, LLC, which he founded in 2010. From 1982 to 2010, Dr. Jawadekar held various technical, management, and business development positions at Pfizer, Inc., including Director, Portfolio Management & Analytics, and Vice President, Asia Colleague Resource Group, for Pfizer Global R&D. Dr. Jawadekar received his B.Pharm. from Shivaji University (1972), M.Pharm. from the University of Bombay (1974), and Ph.D. in Pharmaceutics from the University of Minnesota (1982). Dr. Jawadekar’s academic and professional background in pharmaceuticals provides valuable knowledge and experience to our Board of Directors.
Elona Kogan has served as a Director since June 1, 2018. Ms. Kogan is a biotechnology executive with over 20 years of experience focused on building fast growing publicly traded companies in regulated industries. Ms. Kogan currently serves as the General Counsel & Corporate Secretary of Selecta Biosciences, a clinical stage biopharmaceutical company. Previously, Ms. Kogan served as the General Counsel & Senior Vice President of Government Relations for ARIAD Pharmaceuticals, Inc., a Cambridge, Massachusetts based biotechnology company, from July 2016 through May 2017. Prior to joining ARIAD, Ms. Kogan served as the Vice President of Legal Affairs, and subsequently head of Government Relations, for Avanir Pharmaceuticals, Inc., a California based biotechnology company, during the period of May 2011 through September 2015. Prior roles included positions at King Pharmaceuticals, Inc., Bristol-Meyers Squibb, and Bergen Brunswig Corporation. Ms. Kogan is a graduate of the Southwestern Law School SCALE Program. Ms. Kogan graduated cum laude from Columbia University, Barnard College, with a B.A. degree in Economics. Ms. Kogan’s professional experience working with publicly traded companies in the biotechnology and healthcare arena provides valuable skills and experience to our Board of Directors.
David M. Watumull has served as our Chief Operating Officer since August 2017 and previously as our Vice President, Operations from February 7, 2014 to August 2017. Mr. Watumull has also served as our Assistant Treasurer and Assistant Secretary since February 7, 2014. Mr. Watumull has served as the Chief Operating Officer of Cardax Pharma, Inc. since December 2017 and previously as Vice President, Operations of Cardax Pharma, Inc. from its inception in May 2013 to December 2017. Mr. Watumull has also served as Assistant Treasurer and Assistant Secretary of Cardax Pharma, Inc. since July 2013 and previously as Secretary and Treasurer of Cardax Pharma, Inc. from May 2013 to July 2013. Mr. Watumull also served as Vice President, Operations, Assistant Treasurer, and Assistant Secretary of Cardax Pharmaceuticals, Inc. from July 2013 until it merged with us in December 2015, and previously as Director, Operations and Finance from 2009 to 2013, Operations Manager from 2008 to 2009, and Program Manager from its inception in 2006 to 2009. Mr. Watumull oversees all operations with responsibility for product development and manufacturing, regulatory compliance, sales and marketing, finance, and administration. Mr. Watumull was previously Program Manager at Hawaii Biotech, Inc. from 2005 to 2006, Project Coordinator from 2004 to 2005, and Information Technology Associate / Manager from 2002 to 2004. Mr. Watumull also worked at Aquasearch, Inc., from 2000 to 2001 in various capacities including Medical Information Specialist and Information Technology Associate. Mr. Watumull studied Electrical Engineering at the University of Hawaii.
John B. Russell, CPA has served as our Chief Financial Officer and Treasurer since February 7, 2014. Mr. Russell has served as the Chief Financial Officer and Treasurer of Cardax Pharma, Inc. since July 2013. Mr. Russell also served as the Chief Financial Officer and Treasurer of Cardax Pharmaceuticals, Inc. from July 2013 until it merged with us in December 2015. Mr. Russell is the founder of JBR Business Solutions, LLC and has served as its President since 2010. Mr. Russell has over 20 years of accounting, finance, operations, and SEC reporting experience in biopharmaceutical and high-tech industries. From 2010 to the present, he has served as Chief Financial Officer for various privately-held start-up companies. Mr. Russell was in charge of the Business Advisory Services for the Grant Thornton Honolulu office from 2006 to 2010. From 2005 to 2006, Mr. Russell worked at a consulting company as the Operations Consulting - Financial Management lead, advising Cisco Systems, Inc. Mr. Russell was the General Accounting Manager of the publicly traded company Scios Inc. from 2003 to 2005, where he was in charge of SEC reporting and internal controls. Mr. Russell was the Controller for several portfolio companies in the venture capital firm, Raza Foundries, Inc., from 2001 to 2002, and the General Accounting Manager for inSilicon Corporation, a public company, from 2000 to 2001. Previous to that, Mr. Russell was an auditor at PricewaterhouseCoopers LLP from 1995 to 2000. Mr. Russell is a licensed CPA in Hawaii and has a B.A. in Economics/Accounting from Claremont McKenna College.
Richard M. Morris has served as our Secretary since February 7, 2014. Mr. Morris has served as Secretary of Cardax Pharma, Inc. since December 2017 and previously as Assistant Secretary of Cardax Pharma, Inc. from its inception in May 2013 to December 2017. Mr. Morris also served as Assistant Secretary of Cardax Pharmaceuticals, Inc. from July 2013 until its merger with us in December 2015. Mr. Morris has been a Partner at Allegaert Berger & Vogel LLP, our legal counsel (“ABV”), since November 2018. As a partner of ABV, Mr. Morris represents a variety of clients, primarily in corporate matters. Prior to such position, he was a partner with Herrick, Feinstein LLP since January 2002 and was an associate with such firm since March 1997. Prior to becoming a lawyer, Mr. Morris was an auditor with the Commodities Exchange in New York and later focused on operations and financial management at Kidder Peabody. He also was the U.S. Audit Manager for the financial division for a diversified Australian company. Mr. Morris has a B.S. in Accounting from New York University (1982) and a J.D. from Fordham University School of Law (1990), with bar admissions in New York and Connecticut.
Executive officers are appointed by our Board of Directors. Each executive officer holds his or her office until he or she resigns, is removed by our Board of Directors or his or her successor is elected and qualified. Directors are elected annually by our stockholders at the annual meeting. Each director holds his or her office until his or her successor is elected and qualified or his or her earlier resignation or removal.
| 47 |
Scientific Advisory Board and Key Scientific Personnel
We have assembled a Scientific Advisory Board (“SAB”) and key scientific personnel with expertise in science and medicine with significant experience in pharmaceutical development applicable to our strategy. The members of our SAB and key scientific personnel have made significant scientific contributions in their individual fields, have published in top-tier journals, and have been recognized with numerous awards and distinctions. Our SAB meets on an as-needed basis, based on our need for advice in their respective fields of expertise from time to time. The members of our Scientific Advisory Board and our key scientific personnel are:
| Name | Affiliation, Position | |
| Deepak L. Bhatt, M.D., M.P.H. | Cardax, Chairman of Scientific Advisory Board Harvard Medical School affiliated Brigham and Women’s Hospital, Executive Director of Interventional Cardiovascular Programs Harvard Medical School, Professor | |
| Paresh N. Soni, M.D., Ph.D. | Cardax, Chief Clinical and Regulatory Strategist Cardax, Member of Scientific Advisory Board | |
| R. Preston Mason, Ph.D. | Cardax, Member of Scientific Advisory Board Harvard Medical School-affiliated Brigham and Women’s Hospital, Department of Medicine, Division of Cardiology, Faculty | |
| Gilbert M. Rishton, Ph.D. | Cardax, Chief Science Officer | |
| Jon L. Ruckle, M.D. | Cardax, Chief Medical Officer | |
| Timothy J. King, Ph.D. | Cardax, Vice President, Research |
Deepak L. Bhatt, M.D., M.P.H. has served as Chairman of our Scientific Advisory Board since 2007. Dr. Bhatt has been the Executive Director of Interventional Cardiovascular Programs at Brigham and Women’s Hospital Heart & Vascular Center since December 2013 and Professor of Medicine at Harvard Medical School since June 2012. Dr. Bhatt has authored or co-authored over 1,250 publications and is listed as a Thomson Reuters/Clarivate Analytics Highly Cited Researcher. He is the Editor of Cardiovascular Intervention: A Companion to Braunwald’s Heart Disease and Atherothrombosis in Clinical Practice published by Oxford University Press. He is Senior Associate Editor for News and Clinical Trials for ACC.org. He is the Editor of the peer-reviewed Journal of Invasive Cardiology and Editor-in-Chief of the Harvard Heart Letter for patients. Previously, he was the Chief of Cardiology at VA Boston Healthcare System from 2008 to 2013. He also served as Associate Director of the Cardiovascular Coordinating Center from 2006 to 2008, Associate Director of the Cardiovascular Medicine Fellowship from 2001 to 2005, and Director of the Interventional Cardiology Fellowship from 2002 to 2005 at Cleveland Clinic, where he also served as an interventional cardiologist and an Associate Professor of Medicine. He also completed fellowships in interventional cardiology and cerebral and peripheral vascular intervention and served as Chief Interventional Fellow at Cleveland Clinic. Dr. Bhatt has been listed in Best Doctors in America from 2005 to 2018. He received the Eugene Braunwald Teaching Award for Excellence in the Teaching of Clinical Cardiology from Brigham and Women’s Hospital in 2017 and the ACC’s Distinguished Mentor Award in 2018. Dr. Bhatt’s research interests include acute coronary syndromes, preventive cardiology, and advanced techniques in cardiac, cerebral, and peripheral intervention. Dr. Bhatt obtained his undergraduate science degree as a National Merit Scholar at the Massachusetts Institute of Technology while also serving as a research associate at Harvard Medical School. He received his medical doctorate from Cornell University. His internship and residency in internal medicine were performed at the Hospital of the University of Pennsylvania. Dr. Bhatt also received a Master in Public Health with a concentration in clinical effectiveness from Harvard University.
Paresh N. Soni, M.D., Ph.D. has served as our Chief Clinical and Regulatory Strategist and as a member of Scientific Advisory Board since November 2018. Dr. Soni brings over 20 years of experience working with large and emerging pharmaceutical companies. He has led multidisciplinary teams across the entire drug development spectrum, from translational medicine to successful approval. Dr. Soni serves as a strategic advisor to pharmaceutical and biotechnology companies through his independent consultancy, Soni Biopharma Consulting, LLC, which he founded in September 2018. Previously, Dr. Soni served as Senior Vice President and Head of Development at Amarin Corporation from September 2008 to August 2013, where he led the development and regulatory approval of Vascepa for severe hypertriglyceridemia, and the design and launch of the landmark REDUCE-IT trial for cardiovascular prevention. Prior to joining Amarin, Dr. Soni worked at Pfizer, Inc. from 1999 to 2008, where he held a number of leadership roles in both experimental medicine and late stage development, including the submission of two New Drug Applications. Dr. Soni also served as the Chief Medical Officer of Albireo, a clinical-stage biopharmaceutical company, from September 2016 to August 2018, and as Vice President, Global Medical Sciences and Research at Alexion Pharmaceuticals, from June 2014 to July 2016. Dr. Soni is a member of the American Association for the Study of Liver Diseases. He has authored or co-authored more than 50 scientific papers in peer-reviewed journals, in addition to numerous abstracts. Dr. Soni is a board-certified internist and gastroenterologist. He completed his medical and specialist training at the University of Natal in South Africa. He also completed a research fellowship at the Division of Hepatology, Royal Free Hospital School of Medicine, London, where he received his Ph.D.
R. Preston Mason, Ph.D. has served as a member of Scientific Advisory Board since 2007. Dr. Mason has been on the faculty of the Department of Medicine, Division of Cardiology, at the Harvard Medical School affiliated Brigham and Women’s Hospital since 2002. He is also the President of Elucida Research LLC, a private biotechnology firm he founded in 2001. Previously, he was an associate professor at Drexel University College of Medicine from 1994 to 2001. He served as an assistant professor at the University of Connecticut Health Center from 1989 to 1993. Dr. Mason has published over 250 scientific research articles, book chapters, and abstracts and serves as a reviewer for numerous journals and scientific organizations, including the NIH. He has been the recipient of many awards and patents for his research in cardiovascular pharmacology, including an honorary doctorate in science. Dr. Mason is also a frequent lecturer at national and international meetings. Dr. Mason received his Bachelor of Science degree Summa Cum Laude from Gordon College in 1985. He received his PhD in cell biology and biophysics from the University of Connecticut Health Center in 1989.
Gilbert M. Rishton, Ph.D. has served as our Chief Science Officer since 2009. Dr. Rishton has been the Co-Founder and Chief Chemist at Cognition Therapeutics since January 2007. He was the Founder and Director of the Channel Islands Alzheimer’s Institute, a nonprofit whose mission is to enable new drug development through the identification of high-quality novel drug leads that might become Alzheimer’s disease medicines of the future, from 2004 to 2010. From 1995-2004, he served as a medicinal chemist at Amgen’s Thousand Oaks site, where he was responsible for initiating and building the Amgen Small Molecule Drug Discovery Group, which has grown to become one of the most formidable in the pharmaceutical industry. He also served as the chemistry manager for Amgen’s Sensipar development program, which spanned several phases from preclinical development to manufacturing and then human clinical trials, resulting in the commercial launch of Sensipar, Amgen’s first orally administered small molecule product. He also led the medicinal chemistry program for Amgen’s Secretase Team, which was among the first to produce small molecule secretase inhibitors as potential therapeutic agents for Alzheimer’s disease. Dr. Rishton obtained his undergraduate chemistry degree at University of Rhode Island (1983). He received his Ph.D. degree with a concentration in organic chemistry, organic synthesis, and morphinan synthesis from Florida State University (1988). Dr. Rishton was also a post-doctoral researcher at UC Irvine from 1989-1990.
| 48 |
Jon L. Ruckle, M.D. has acted as our Chief Medical Officer and in related medical advisory roles for us since 2013. Dr. Ruckle is a physician with over 20 years of full-time experience in clinical pharmacology research as an Investigator (over 350 studies), Medical Director of clinical research units devoted to Phase I studies, and consultant. Consulting activities focus on clinical study design for first-in-human through proof of concept studies, protocol development and writing, and medical monitoring. As Principal of Pacific Pharma Group, LLC, which Dr. Ruckle founded in 2008, he provides consultation services for the pharmaceutical and nutraceutical industry, including study design, product development strategy, and medical monitoring. Dr. Ruckle served as the founding Medical Director at Northwest Kinetics in Tacoma, WA, 1995 to 2000 (later acquired by Charles River Labs and subsequently by Comprehensive Clinical Development), then led Phase I development at Radiant Research Honolulu from 2000 until acquired by Covance in 2006, remained as Medical Director at Covance Honolulu to 2008, then founded Pacific Pharma Group to provide consulting services, and also served as Medical Director, Early Development for Comprehensive Clinical Development in Tacoma WA from 2011 to 2013.
Timothy J. King, Ph.D. has served as our Vice President, Research since 2009 and previously as our Senior Director of Biological Research from 2007 to 2009 and Director of Biological Research from 2006 to 2007. Dr. King is an expert on the mechanism of action and biological applications of astaxanthin and related carotenoids. Dr. King was the Director of Cancer Chemoprevention at Hawaii Biotech, Inc. from 2005-2006. From 2003-2005, he served as a Staff Scientist at the Fred Hutchinson Cancer Research Center, where he also served as a post-doctoral researcher from 1999 to 2004. Dr. King has over 25 years of combined academic and private sector scientific research experience including utilizing cell culture and animal model systems to address a wide range of topics including cardiovascular disease, liver disease, thrombosis, and cancer. Dr. King received his undergraduate degree in Biochemistry/Cell Biology from University of California San Diego. He obtained his Master of Science in biology (Molecular Virology) from San Diego State University studying Rhadoviral transcription processes in 1993. In 1999, he obtained his Ph.D. in Genetics/Molecular Biology from the University of Hawaii at Manoa where he focused on the tumor suppressor and growth regulatory roles of gap junction proteins using cancer cell culture systems and mouse tumor models. Concurrently, he also studied the influence of retinoids and various carotenoids on normal and tumor cell growth/behavior, carcinogenesis and gene regulation.
Family Relationships
David G. Watumull is the father of David M. Watumull. There are no other family relationships among any of our officers or directors.
Involvement in Certain Legal Proceedings
To the best of our knowledge, none of our directors or executive officers have been convicted in a criminal proceeding, excluding traffic violations or similar misdemeanors, or has been a party to any judicial or administrative proceeding during the past ten years that resulted in a judgment, decree, or final order enjoining the person from future violations of, or prohibiting activities subject to, federal or state securities laws, or a finding of any violation of federal or state securities laws, except for matters that were dismissed without sanction or settlement. Except as set forth in our discussion below in “Certain Relationships and Related Transactions” none of our directors, director nominees, or executive officers has been involved in any transactions with us or any of our directors, executive officers, affiliates, or associates which are required to be disclosed pursuant to the rules and regulations of the Commission.
Code of Ethics
Our Code of Business Conduct and Ethics, effective as of February 7, 2014 (the “Code of Ethics”), contains the ethical principles by which our Chief Executive Officer and Chief Financial Officer, among others, are expected to conduct themselves when carrying out their duties and responsibilities. A copy of our Code of Ethics may be found on our website at www.cardaxpharma.com. We will provide a copy of our Code of Ethics to any person, without charge, upon request, by writing to David G. Watumull, Cardax, Inc., 2800 Woodlawn Drive, Suite 129, Honolulu, Hawaii 96822.
Board Committees
We are not required under the Securities and Exchange Act to maintain any committees of our Board of Directors. We have formed certain committees of our board as a matter of preferred corporate practices.
We have an audit committee, a compensation committee and a nominating and corporate governance committee, each of which has the composition and responsibilities described below.
Audit Committee. Our audit committee oversees a broad range of issues surrounding our accounting and financial reporting processes and audits of our consolidated financial statements, including the following:
| ● | monitors the integrity of our financial statements, our compliance with legal and regulatory requirements, our independent registered public accounting firm’s qualifications and independence, and the performance of our internal audit function and independent registered public accounting firm; | |
| ● | assumes direct responsibility for the appointment, compensation, retention and oversight of the work of any independent registered public accounting firm engaged for the purpose of performing any audit, review or attest services and for dealing directly with any such accounting firm; | |
| ● | provides a medium for consideration of matters relating to any audit issues; and | |
| ● | prepares the audit committee report that the rules require be included in our filings with the SEC. |
| 49 |
The members of our audit committee are Terence A. Kelly, Ph.D. (Chairperson), Makarand Jawadekar, Ph.D., and Elona Kogan. Effective at the effective date of this prospectus, George W. Bickerstaff, III will join the audit committee.
Our Board of Directors has determined that each member of the audit committee meets the requirements for independence and financial literacy under the applicable rules and regulations of the SEC and the listing standards of the Nasdaq Capital Market and has also determined that Mr. Bickerstaff is an “audit committee financial expert” as defined in the rules of the SEC and has the requisite financial sophistication as defined under the listing standards of the Nasdaq Capital Market based on his prior position as the former Chief Financial Officer of Novartis Pharma AG, one of the largest pharmaceutical companies in the world, and his other senior financial positions.
The Company’s audit committee operates under a written charter which satisfies the applicable rules and regulations of the SEC and the listing standards of the Nasdaq Capital Market, which charter is available on our website at www.cardaxpharma.com.
Compensation Committee. Our compensation committee reviews and recommends policy relating to compensation and benefits of our officers, directors and employees, including reviewing and approving corporate goals and objectives relevant to the compensation of our Chief Executive Officer and other senior officers, evaluating the performance of these persons in light of those goals and objectives and setting compensation of these persons based on such evaluations. The compensation committee reviews and evaluates, at least annually, the performance of the compensation committee and its members, including compliance of the compensation committee with its charter.
The members of our compensation committee are Elona Kogan (Chairperson), Makarand Jawadekar, Ph.D., and Michele Galen. Our compensation committee operates under a written charter which satisfies the applicable rules and regulations of the SEC and the listing standards of the Nasdaq Capital Market, which charter is available on our website at www.cardaxpharma.com.
Nominating and Corporate Governance Committee. The nominating and corporate governance committee oversees and assists our Board of Directors in identifying, reviewing and recommending nominees for election as directors; evaluating our Board of Directors and our management; developing, reviewing and recommending corporate governance guidelines and a corporate code of business conduct and ethics; and generally advises our Board of Directors on corporate governance and related matters.
The members of our nominating and corporate governance committee are Michele Galen (Chairperson), Makarand Jawadekar, Ph.D., and Terence A. Kelly, Ph.D. Our nominating and corporate governance committee has a written charter available on our website at www.cardaxpharma.com.
Director Independence
George W. Bickerstaff, III, Terence A. Kelly, Ph.D., Michele Galen, Makarand Jawadekar, Ph.D., and Elona Kogan are our independent directors, based on the definition of “independence” of The Nasdaq Stock Market. Nasdaq Listing Rule 5605(a)(2) provides that an “independent director” is a person other than an officer or employee of the Company or any other individual having a relationship that, in the opinion of the Company’s Board of Directors, would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. The Nasdaq listing rules provide that a director cannot be considered independent if:
| ● | the director is, or at any time during the past three years was, an employee of the Company; | |
| ● | the director or a family member of the director accepted any compensation from the Company in excess of $120,000 during any period of 12 consecutive months within the three years preceding the independence determination (subject to certain exclusions, including, among other things, compensation for board or board committee service); | |
| ● | a family member of the director is, or at any time during the past three years was, an executive officer of the Company; | |
| ● | the director or a family member of the director is a partner in, controlling stockholder of, or an executive officer of an entity to which the Company made, or from which the Company received, payments in the current or any of the past three fiscal years that exceed 5% of the recipient’s consolidated gross revenue for that year or $200,000, whichever is greater (subject to certain exclusions); | |
| ● | the director or a family member of the director is employed as an executive officer of an entity where, at any time during the past three years, any of the executive officers of the Company served on the compensation committee of such other entity; or | |
| ● | the director or a family member of the director is a current partner of the Company’s outside auditor, or at any time during the past three years was a partner or employee of the Company’s outside auditor, and who worked on the Company’s audit. |
Indemnification
We maintain directors’ and officers’ liability insurance. Our amended and restated certificate of incorporation and amended and restated bylaws include provisions limiting the liability of directors and officers and indemnifying them under certain circumstances. We have entered into indemnification agreements with our directors to provide our directors and certain of their affiliated parties with additional indemnification and related rights. See “Indemnification of Directors and Officers” for further information.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers or persons controlling the Company pursuant to Delaware law, we are informed that in the opinion of the Securities and Exchange Commission, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
| 50 |
Conflicts of Interest
Certain potential conflicts of interest are inherent in the relationships between our officers and directors and us.
From time to time, one or more of our affiliates may form or hold an ownership interest in and/or manage other businesses both related and unrelated to the type of business that we own and operate. These persons expect to continue to form, hold an ownership interest in and/or manage additional other businesses which may compete with our business with respect to operations, including financing and marketing, management time and services and potential customers. These activities may give rise to conflicts between or among the interests of us and other businesses with which our affiliates are associated. Our affiliates are in no way prohibited from undertaking such activities, and neither us nor our stockholders will have any right to require participation in such other activities.
Further, because we intend to transact business with some of our officers, directors and affiliates, as well as with firms in which some of our officers, directors or affiliates have a material interest, potential conflicts may arise between the respective interests of us and these related persons or entities. We believe that such transactions will be effected on terms at least as favorable to us as those available from unrelated third parties.
With respect to transactions involving real or apparent conflicts of interest, we have adopted policies and procedures which require that: (i) the fact of the relationship or interest giving rise to the potential conflict be disclosed or known to the directors who authorize or approve the transaction prior to such authorization or approval; and (ii) the transaction be fair and reasonable to us at the time it is authorized or approved by our directors.
COMPENSATION OF EXECUTIVE OFFICERS AND DIRECTORS
Compensation of Executive Officers
The following sets forth information with respect to the compensation awarded or paid to David G. Watumull, our Chief Executive Officer, and David M. Watumull, our Chief Operating Officer, for all services rendered in all capacities to the Company and its predecessors during the fiscal years ending December 31, 2017 and 2018. These executive officers are referred to as the “named executive officers” throughout this prospectus. In addition, the following sets forth information with respect to the compensation awarded or paid to our two highest compensated individuals not serving as executive officers, Gilbert M. Rishton, our Chief Science Officer, and Timothy J. King, our Vice President of Research, for all services rendered in all capacities to the Company and its predecessors during the fiscal years ending December 31, 2017 and 2018.
The following table sets forth information regarding each element of compensation provided to our named executive officers, and our two highest compensated individuals not serving as executive officers, for the fiscal years ended December 31, 2017 and 2018:
| Name | Year | Salary(1) | All
Other Comp.(2) | Total | ||||||||||
| David G. Watumull | 2017 | $ | 138,461 | (3) | $ | 9,222 | $ | 147,683 | ||||||
| Chief Executive Officer | 2018 | $ | 187,500 | (3) | $ | 10,534 | $ | 198,034 | ||||||
| David M. Watumull | 2017 | $ | 107,500 | (4) | $ | 7,350 | $ | 114,850 | ||||||
| Chief Operating Officer | 2018 | $ | 150,000 | (4) | $ | 7,443 | $ | 157,443 | ||||||
| Gilbert M. Rishton | 2017 | $ | 76,827 | (5) | $ | 525 | $ | 77,352 | ||||||
| Chief Science Officer | 2018 | $ | 127,500 | (5) | $ | 1,058 | $ | 128,558 | ||||||
| Timothy J. King | 2017 | $ | 99,712 | (6) | $ | - | $ | 99,712 | ||||||
| Vice President, Research | 2018 | $ | 127,500 | (6) | $ | 11,500 | $ | 139,000 | ||||||
| (1) | The amounts disclosed refer to salary (paid in cash). | |
| (2) | The amounts disclosed refer to (i) imputed income in connection with certain benefits and/or insurance premiums paid in lieu of additional cash compensation, or (ii) other cash compensation. | |
| (3) | As of January 1, 2017, Mr. David G. Watumull received bi-weekly compensation equal to $4,327. On August 31, 2017, the compensation arrangement of Mr. David G. Watumull was amended so that, effective September 1, 2017, he received bi-weekly compensation equal to $7,212. | |
| (4) | As of January 1, 2017, Mr. David M. Watumull received bi-weekly compensation equal to $3,269. On August 31, 2017, the compensation arrangement of Mr. David M. Watumull was amended so that, effective September 1, 2017, he received bi-weekly compensation equal to $5,769. | |
| (5) | As of January 1, 2017, Mr. Rishton received bi-weekly compensation equal to $1,923. On August 31, 2017, the compensation arrangement of Mr. Rishton was amended so that, effective September 1, 2017, he received bi-weekly compensation equal to $4,904. | |
| (6) | As of January 1, 2017, Mr. King received bi-weekly compensation equal to $3,269. On August 31, 2017, the compensation arrangement of Mr. King was amended so that, effective September 1, 2017, he received bi-weekly compensation equal to $4,904. |
| 51 |
Outstanding Equity Awards to Executive Officers at Fiscal Year-End 2018
The following table sets forth information regarding outstanding option awards to our named executive officers as of December 31, 2018:
| Option awards(1)(2) | ||||||||||||||||||
| Name | Number
of securities underlying unexercised options exercisable | Number
of securities underlying unexercised options unexercisable | Number
of securities underlying unexercised unearned options | Option exercise price ($) | Option
expiration date | |||||||||||||
| David G. Watumull | 1,750,588 | - | - | $ | 0.155 | February 7, 2024 | ||||||||||||
| David G. Watumull | 4,941,845 | - | - | $ | 0.625 | February 7, 2024 | ||||||||||||
| David G. Watumull | 468,498 | (3) | - | - | $ | 0.32 | June 30, 2020 | |||||||||||
| David G. Watumull | 390,686 | (3) | - | - | $ | 0.20 | June 30, 2020 | |||||||||||
| David G. Watumull | 89,523 | (3) | - | - | $ | 0.49 | September 30, 2020 | |||||||||||
| David G. Watumull | 137,675 | (3) | - | - | $ | 0.27 | December 31, 2020 | |||||||||||
| David G. Watumull | 774,385 | (3) | - | - | $ | 0.06 | March 31, 2021 | |||||||||||
| David M. Watumull | 45,058 | - | - | $ | 0.155 | February 7, 2024 | ||||||||||||
| David M. Watumull | 2,388,554 | - | - | $ | 0.625 | February 7, 2024 | ||||||||||||
| David M. Watumull | 160,806 | (3) | - | - | $ | 0.32 | June 30, 2020 | |||||||||||
| David M. Watumull | 284,917 | (3) | - | - | $ | 0.20 | June 30, 2020 | |||||||||||
| David M. Watumull | 67,639 | (3) | - | - | $ | 0.49 | September 30, 2020 | |||||||||||
| David M. Watumull | 104,021 | (3) | - | - | $ | 0.27 | December 31, 2020 | |||||||||||
| David M. Watumull | 562,846 | (3) | - | - | $ | 0.06 | March 31, 2021 | |||||||||||
| (1) | The type of securities underlying all outstanding option awards is our common stock. | |
| (2) | None of our named executive officers have received stock awards. | |
| (3) | Stock options awarded in lieu of cash compensation. |
Compensation of Directors
The following table sets forth information regarding each element of compensation that we paid or awarded to our independent directors for the fiscal years ended December 31, 2017 and 2018:
| Name | Year | Cash Comp. | Equity Awards | Total(1) | ||||||||||
| George W. Bickerstaff, III | 2017 | $ | - | $ | 58,333 | $ | 58,333 | |||||||
| George W. Bickerstaff, III | 2018 | $ | - | $ | 75,000 | $ | 75,000 | |||||||
| Terence A. Kelly, Ph.D. | 2017 | $ | - | $ | 58,333 | $ | 58,333 | |||||||
| Terence A. Kelly, Ph.D. | 2018 | $ | 25,000 | $ | 50,000 | $ | 75,000 | |||||||
| Michele Galen(2) | 2017 | $ | - | $ | 58,333 | $ | 58,333 | |||||||
| Michele Galen | 2018 | $ | - | $ | 75,000 | $ | 75,000 | |||||||
| Makarand Jawadekar, Ph.D. (3) | 2018 | $ | - | $ | 43,750 | $ | 43,750 | |||||||
| Elona Kogan(4) | 2018 | $ | - | $ | 43,750 | $ | 43,750 | |||||||
| (1) | The amounts disclosed represent compensation in connection with services provided by each independent director. As of January 1, 2017, each independent director received quarterly compensation of $12,500 in arrears. On August 31, 2017, the compensation arrangement was amended so that effective September 1, 2017, each independent director received quarterly compensation of $18,750 in arrears. Independent director compensation is payable in the form of a grant of shares of our common stock or non-qualified stock options to purchase shares of our common stock based on the higher of the then current market price or $0.15 per share, with up to one-third payable in cash at the election of the director. |
| (3) | Ms. Galen was elected to the Board of Directors on January 4, 2017. |
| (3) | Dr. Jawadekar was elected to the Board of Directors on June 1, 2018. |
| (3) | Ms. Kogan was elected to the Board of Directors on June 1, 2018. |
| 52 |
Outstanding Equity Awards to Directors at Fiscal Year-End 2018
The following table sets forth information regarding outstanding equity awards to our independent directors as of December 31, 2018:
| Stock awards(1) | Option awards (2) | |||||||||||||||||||||
| Name | Number
of securities awarded | Number
of securities underlying unexercised options exercisable | Number
of securities underlying unexercised options unexercisable | Number
of securities underlying unexercised unearned options | Option exercise price ($) | Option
expiration date | ||||||||||||||||
| George W. Bickerstaff, III | 1,555,896 | - | - | - | $ | - | - | |||||||||||||||
| Terence A. Kelly, Ph.D. | 795,980 | - | - | - | $ | - | - | |||||||||||||||
| Terence A. Kelly, Ph.D. | - | 416,667 | - | - | $ | 0.06 | March 31, 2021 | |||||||||||||||
| Terence A. Kelly, Ph.D. | - | 27,778 | - | - | $ | 0.15 | September 30, 2021 | |||||||||||||||
| Terence A. Kelly, Ph.D. | - | 83,333 | - | - | $ | 0.15 | December 31, 2021 | |||||||||||||||
| Terence A. Kelly, Ph.D. | - | 78,125 | - | - | $ | 0.185 | March 31, 2022 | |||||||||||||||
| Terence A. Kelly, Ph.D. | - | 83,333 | - | - | $ | 0.20 | June 30, 2022 | |||||||||||||||
| Michele Galen | 660,332 | - | - | - | $ | - | - | |||||||||||||||
| Makarand Jawadekar, Ph.D. | 215,909 | - | - | - | $ | - | - | |||||||||||||||
| Elona Kogan | 215,909 | - | - | - | $ | - | - | |||||||||||||||
| (1) | All shares are fully vested. | |
| (2) | The type of securities underlying all outstanding option awards is our common stock. |
Employment and Consulting Agreements
Executive Officer Compensation
On February 7, 2014, we entered into employment agreements with each of Messrs. David G. Watumull, David M. Watumull, Gilbert M. Rishton, and Timothy J. King, which provided for employment for an initial term of one year, subject to renewal and earlier termination rights as provided in such agreements. These agreements provide for compensation terms and duration of employment as set forth in each such agreement. Such agreements include restrictive covenants concerning competition with us and solicitation of our employees and clients, if such individuals are terminated for cause as defined in such agreements.
| ● | To conserve cash resources while seeking additional financing, we and our employees, including Messrs. David G. Watumull, David M. Watumull, Gilbert M. Rishton, and Timothy J. King, agreed to reduce cash compensation effective January 15, 2015. | |
| ● | On June 30, 2015, the compensation arrangements of Messrs. David G. Watumull, David M. Watumull, Gilbert M. Rishton, and Timothy J. King were amended so that, effective after June 30, 2015, we had the right to pay any compensation due to such officer during any calendar quarter that was not paid in cash in the form of shares of our common stock or incentive stock options under the 2014 Plan. In addition, the amount of the unpaid cash compensation that accrued during the first and second quarters of 2015 was paid with incentive stock options under the 2014 Plan. | |
| ● | On March 28, 2016, we furloughed all of our employees and independent contractors indefinitely and arranged with our Chief Executive Officer, David G. Watumull; our Chief Financial Officer, John B. Russell; and our Vice President, Operations, David M. Watumull, to continue their services for cash compensation equal to the minimum wage. In addition, each of the directors agreed, effective April 1, 2016, to suspend any additional equity compensation, until otherwise agreed by the Company. | |
| ● | On June 3, 2016, the compensation arrangement of David M. Watumull was amended so that, effective May 30, 2016, he would receive bi-weekly compensation equal to $3,269 and the compensation arrangement of Timothy J. King was amended so that, effective May 30, 2016, he would receive bi-weekly compensation equal to $1,635. | |
| ● | On September 6, 2016, the compensation arrangements of certain officers were amended so that effective September 8, 2016, (i) David G. Watumull would receive bi-weekly compensation equal to $4,327, (ii) Gilbert M. Rishton would receive bi-weekly compensation equal to $1,923, and (iii) Timothy J. King would receive bi-weekly compensation equal to $3,269. | |
| ● | On August 31, 2017, the compensation arrangements of certain officers were amended so that effective September 1, 2017, (i) David G. Watumull would receive bi-weekly compensation equal to $7,212, (ii) David M. Watumull would receive bi-weekly compensation equal to $5,769, (iii) Gilbert M. Rishton would receive bi-weekly compensation equal to $4,904, and (iv) Timothy J. King would receive bi-weekly compensation equal to $4,904. |
| 53 |
On July 30, 2013, we entered into a service agreement with JBR Business Solutions, LLC, under which John B. Russell agreed to serve as our Chief Financial Officer, and under which Mr. Russell would be paid an aggregate of $7,000 a month. Mr. Russell is the Managing Partner of JBR Business Solutions, LLC. To conserve cash resources while seeking additional financing, we and Mr. Russell, agreed to reduce cash compensation effective January 15, 2015. On June 30, 2015, the compensation arrangement was amended so that, effective after June 30, 2015, we had the right to pay up to 50% of any compensation due during any calendar quarter that was not paid in cash in the form of shares of our common stock or non-qualified stock options under the 2014 Plan. On March 28, 2016, Mr. Russell was furloughed and agreed to continue service as Chief Financial Officer for cash compensation equal to the minimum wage. On September 6, 2016, the compensation arrangement was amended so that effective September 30, 2016, he would receive monthly compensation of $3,500. On August 31, 2017, the compensation arrangement was amended so that effective September 1, 2017, Mr. Russell would receive monthly compensation of $5,250.
Director Compensation
On June 30, 2015, we entered into an agreement with George W. Bickerstaff, III and Terence A. Kelly, Ph.D. that provided for the annual compensation of each independent director equal to $100,000, payable quarterly in arrears in the form of a grant of shares of our common stock or non-qualified stock options to purchase shares of our common stock under the 2014 Plan.
Effective April 1, 2016, the independent directors of the Company agreed to suspend any additional equity compensation, until otherwise agreed by the Company
On September 6, 2016, the compensation arrangements of the independent directors of the Company were amended so that effective September 30, 2016, they would each receive quarterly equity compensation of $12,500 in arrears in the form of a grant of shares of our common stock or non-qualified stock options to purchase shares of our common stock under the 2014 Plan based on the higher of the then current market price or $0.15 per share, with such compensation prorated for one of three months for the quarter ended September 30, 2016.
On January 4, 2017, our Board of Directors elected Michele Galen to serve as an independent director until our next annual meeting of stockholders with quarterly equity compensation of $12,500 in arrears in the form of a grant of shares of our common stock or non-qualified stock options to purchase shares of our common stock under the 2014 Plan based on the higher of the then current market price or $0.15 per share.
On August 31, 2017, the compensation arrangements of the independent directors of the Company were amended so that effective September 1, 2017, they would each receive quarterly equity compensation of $18,750 in arrears in the form of a grant of shares of our common stock or non-qualified stock options to purchase shares of our common stock under the 2014 Plan based on the higher of the then current market price or $0.15 per share. In 2018, independent director compensation was updated such that up to one-third could be payable in cash at the election of the director.
On June 1, 2018, our Board of Directors elected Makarand Jawadekar, Ph.D. and Elona Kogan to serve as independent directors until our next annual meeting of stockholders with quarterly compensation of $18,750 in arrears in the form of a grant of shares of our common stock or non-qualified stock options to purchase shares of our common stock under the 2014 Plan based on the higher of the then current market price or $0.15 per share, with up to one-third payable in cash at the election of the director. Dr. Jawadekar’s and Ms. Kogan’s compensation for the second quarter of 2018 was prorated to $6,250.
2014 Equity Compensation Plan
Our 2014 Plan is administered by our compensation committee. The purpose of the 2014 Plan is to provide financial incentives for selected directors, employees, advisers, and consultants of Cardax and/or its subsidiaries, thereby promoting the long-term growth and financial success of the Company. The issuance of awards under the 2014 Plan is at the discretion of our compensation committee, which has the authority to determine the persons to whom any awards shall be granted and the terms, conditions, and restrictions applicable to any award. Under the 2014 Plan, we may grant equity-based incentive awards, including options, restricted stock, and other stock-based awards, to any directors, employees, advisers, and consultants that provide services to us or any of our subsidiaries. The 2014 Plan also permits us to amend the terms of previously granted options or other awards. An aggregate of 50,420,148 shares of our common stock have been reserved for issuance under the 2014 Plan, which is subject to adjustment as described in such plan. On December 4, 2018, our stockholders and our Board of Directors authorized the annual increase of the Plan Shares on January 1st of each year, at the discretion of our Board of Directors, by up to such number of shares that is equal to four percent (4%) of the shares of our common stock issued and outstanding as of December 31st of the previous calendar year. As of the date of this prospectus, there are 3,256,709 shares of our common stock available for future awards under the 2014 Plan.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth information regarding the ownership of our common stock as of November 20, 2019 for:
| ● | each director; | |
| ● | each person known by us to own beneficially 5% or more of our common stock; | |
| ● | each officer named in the summary compensation table elsewhere in this prospectus; and | |
| ● | all directors and executive officers as a group. |
The amounts and percentages of our common stock beneficially owned are reported on the basis of regulations of the SEC governing the determination of beneficial ownership of securities. Under the rules of the SEC, a person is deemed to be a “beneficial owner” of a security if that person has or shares “voting power,” which includes the power to vote or to direct the voting of such security, or “investment power,” which includes the power to dispose of or to direct the disposition of such security. A person is also deemed to be a beneficial owner of any securities of which that person has the right to acquire beneficial ownership within 60 days. Under these rules more than one person may be deemed a beneficial owner of the same securities and a person may be deemed to be a beneficial owner of securities as to which such person has no economic interest.
| 54 |
Unless otherwise indicated below, to our knowledge each beneficial owner named in the table has sole voting and sole investment power with respect to all shares beneficially owned, subject to community property laws where applicable.
| Amount of Beneficial Ownership of our Common Stock | ||||||||||||
| Name | Number of Shares | %(1) | %(2) | |||||||||
| Directors and Executive Officers | ||||||||||||
| George W. Bickerstaff, III(3) | 4,621,772 | (4) | 3.4 | % | ||||||||
| Terence A. Kelly, Ph.D.(5) | 1,717,671 | (6) | 1.2 | |||||||||
| Michele Galen(7) | 1,009,016 | (8) | * | |||||||||
| Makarand Jawadekar, Ph.D.(9) | 1,085,169 | (10) | * | |||||||||
| Elona Kogan(11) | 564,593 | (12) | * | |||||||||
| David G. Watumull(13) | 11,351,372 | (14) | 7.7 | % | ||||||||
| David M. Watumull(15) | 3,613,841 | (16) | 2.6 | % | ||||||||
| John B. Russell(17) | 331,997 | (18) | * | % | ||||||||
| All directors and executive officers as a group (6 persons) | 24,295,431 | 15.9 | % | |||||||||
| Beneficial Owner of 5% or more | ||||||||||||
| Eric J. Pearson and Lianne L. Pearson(19) | 44,291,589 | (20) | 28.1 | % | ||||||||
| James K. Schuler(21) | 31,436,420 | (22) | 19.4 | % | ||||||||
| * | Represents beneficial ownership of our common stock of less than 1%. |
| (1) | Based on 137,261,594 shares of our common stock issued and outstanding as of November 20, 2019. |
| (2) | Based on shares of our common stock issued and outstanding after giving effect to the shares of common stock issued in this offering. |
| (3) | The address of Mr. George W. Bickerstaff, III is c/o Cardax, Inc., 2800 Woodlawn Drive, Honolulu, Hawaii 96822. Mr. Bickerstaff is the current Chairman of our Board of Directors. |
| (4) | Represents 4,621,772 shares of our common stock. |
| (5) | The address of Dr. Terence A. Kelly is c/o Cardax, Inc., 2800 Woodlawn Drive, Honolulu, Hawaii 96822. Dr. Kelly is a member of our Board of Directors. |
| (6) | Represents (a) 1,028,435 shares of our common stock, (b) 416,667 shares of our common stock issuable upon exercise by Dr. Kelly of options that are presently exercisable, at an exercise price of $0.06 per share, (c) 111,111 shares of our common stock issuable upon exercise by Dr. Kelly of options that are presently exercisable, at an exercise price of $0.15 per share, (d) 78,125 shares of our common stock issuable upon exercise by Dr. Kelly of options that are presently exercisable, at an exercise price of $0.185 per share, and (e) 83,333 shares of our common stock issuable upon exercise by Dr. Kelly of options that are presently exercisable, at an exercise price of $0.20 per share. |
| (7) | The address of Ms. Michele Galen is c/o Cardax, Inc., 2800 Woodlawn Drive, Honolulu, Hawaii 96822. Ms. Galen is a member of our Board of Directors. |
| (8) | Represents 1,009,016 shares of our common stock. |
| (9) | The address of Dr. Makarand Jawadekar, Ph.D. is c/o Cardax, Inc., 2800 Woodlawn Drive, Honolulu, Hawaii 96822. Dr. Jawadekar is a member of our Board of Directors. |
| (10) | Represents (a) 564,593 shares of our common stock, (b) 95,238 shares of our common stock issuable upon exercise of options that are presently exercisable, at an exercise price of $0.32 per share, which Dr. Jawadekar may be deemed to beneficially own as the principal of Melinda Consulting, LLC, (c) 115,385 shares of our common stock issuable upon exercise of options that are presently exercisable, at an exercise price of $0.20 per share, which Dr. Jawadekar may be deemed to beneficially own as the principal of Melinda Consulting, LLC, (d) 25,862 shares of our common stock issuable upon exercise of options that are presently exercisable, at an exercise price of $0.49 per share, which Dr. Jawadekar may be deemed to beneficially own as the principal of Melinda Consulting, LLC, (e) 34,091 shares of our common stock issuable upon exercise of options that are presently exercisable, at an exercise price of $0.27 per share, which Dr. Jawadekar may be deemed to beneficially own as the principal of Melinda Consulting, LLC, and (f) 250,000 shares of our common stock issuable upon exercise of options that are presently exercisable, at an exercise price of $0.06 per share, which Dr. Jawadekar may be deemed to beneficially own as the principal of Melinda Consulting, LLC. |
| 55 |
| (11) | The address of Ms. Elona Kogan is c/o Cardax, Inc., 2800 Woodlawn Drive, Honolulu, Hawaii 96822. Ms. Kogan is a member of our Board of Directors. |
| (12) | Represents 564,593 shares of our common stock. |
| (13) | The address of Mr. David G. Watumull is c/o Cardax, Inc., 2800 Woodlawn Drive, Honolulu, Hawaii 96822. Mr. David G. Watumull is our President, CEO, and a member of our Board of Directors. |
| (14) | Represents (a) 1,750,588 shares of our common stock issuable upon exercise by Mr. David G. Watumull of options that are presently exercisable, at an exercise price of $0.155 per share, (b) 4,941,845 shares of our common stock issuable upon exercise by Mr. David G. Watumull of options that are presently exercisable, at an exercise price of $0.625 per share, (c) 468,498 shares of our common stock issuable upon exercise by Mr. David G. Watumull of options that are presently exercisable, at an exercise price of $0.32 per share, (d) 390,686 shares of our common stock issuable upon exercise by Mr. David G. Watumull of options that are presently exercisable, at an exercise price of $0.20 per share, (e) 89,523 shares of our common stock issuable upon exercise by Mr. David G. Watumull of options that are presently exercisable, at an exercise price of $0.49 per share, (f) 137,675 shares of our common stock issuable upon exercise by Mr. David G. Watumull of options that are presently exercisable, at an exercise price of $0.27 per share, (g) 774,385 shares of our common stock issuable upon exercise by Mr. David G. Watumull of options that are presently exercisable, at an exercise price of $0.06 per share, (h) 398,172 shares of our common stock issued in the Holdings Merger, which Mr. Watumull may be deemed to beneficially own as the Trustee of the David G. Watumull Revocable Living Trust, (i) 350,000 shares of our common stock issued in the 2016/2017 Unit Offering, which Mr. Watumull may be deemed to beneficially own as the Trustee of the David G. Watumull Revocable Living Trust, (j) 350,000 shares of our common stock issuable upon exercise of a warrant issued in the 2016/2017 Unit Offering at an exercise price of $0.08 per share, which Mr. Watumull may be deemed to beneficially own as the Trustee of the David G. Watumull Revocable Living Trust, (k) 350,000 shares of our common stock issuable upon exercise of a warrant issued in the 2016/2017 Unit Offering at an exercise price of $0.12 per share, which Mr. Watumull may be deemed to beneficially own as the Trustee of the David G. Watumull Revocable Living Trust, (l) 350,000 shares of our common stock issuable upon exercise of a warrant issued in the 2016/2017 Unit Offering at an exercise price of $0.16 per share, which Mr. Watumull may be deemed to beneficially own as the Trustee of the David G. Watumull Revocable Living Trust, and (m) 1,000,000 shares of our common stock issuable upon conversion of a convertible note at an exercise price of $0.10 per share. |
| (15) | The address of Mr. David M. Watumull is c/o Cardax, Inc., 2800 Woodlawn Drive, Honolulu, Hawaii 96822. Mr. David M. Watumull is our Chief Operating Officer. |
| (16) | Represents (a) 45,058 shares of our common stock issuable upon exercise by Mr. David M. Watumull of options that are presently exercisable, at an exercise price of $0.155 per share, (b) 2,388,554 shares of our common stock issuable upon exercise by Mr. David M. Watumull of options that are presently exercisable, at an exercise price of $0.625 per share, (c) 160,806 shares of our common stock issuable upon exercise by Mr. David M. Watumull of options that are presently exercisable, at an exercise price of $0.32 per share, (d) 284,917 shares of our common stock issuable upon exercise by Mr. David M. Watumull of options that are presently exercisable, at an exercise price of $0.20 per share, (e) 67,639 shares of our common stock issuable upon exercise by Mr. David M. Watumull of options that are presently exercisable, at an exercise price of $0.49 per share, (f) 104,021 shares of our common stock issuable upon exercise by Mr. David M. Watumull of options that are presently exercisable, at an exercise price of $0.27 per share, and (g) 562,846 shares of our common stock issuable upon exercise by Mr. David M. Watumull of options that are presently exercisable, at an exercise price of $0.06 per share. |
| (17) | The address of Mr. John B. Russell is c/o Cardax, Inc., 2800 Woodlawn Drive, Honolulu, Hawaii 96822. Mr. Russell is our Chief Financial Officer. |
| (18) | Represents (a) 59,835 shares of our common stock issuable upon exercise of options that are presently exercisable, at an exercise price of $0.32 per share, which Mr. Russell may be deemed to beneficially own as the Managing Partner of JBR Business Solutions, LLC, (b) 62,424 shares of our common stock issuable upon exercise of options that are presently exercisable, at an exercise price of $0.20 per share, which Mr. Russell may be deemed to beneficially own as the Managing Partner of JBR Business Solutions, LLC, (c) 18,956 shares of our common stock issuable upon exercise of options that are presently exercisable, at an exercise price of $0.49 per share, which Mr. Russell may be deemed to beneficially own as the Managing Partner of JBR Business Solutions, LLC, (d) 24,988 shares of our common stock issuable upon exercise of options that are presently exercisable, at an exercise price of $0.27 per share, which Mr. Russell may be deemed to beneficially own as the Managing Partner of JBR Business Solutions, LLC, and (e) 165,794 shares of our common stock issuable upon exercise of options that are presently exercisable, at an exercise price of $0.06 per share, which Mr. Russell may be deemed to beneficially own as the Managing Partner of JBR Business Solutions, LLC. |
| (19) | The address of Dr. Eric J. Pearson and Mrs. Lianne L. Pearson is 814 Mokulua Drive, Kailua, Hawaii 96734. Dr. and Mrs. Pearson do not have any position, office, contractual relationship, or other understanding with the Company regarding the management or control of the Company and accordingly, we have determined that such stockholder is not an affiliate of Cardax. |
| 56 |
| (20) | Represents (a) 208,333 shares of our common stock issued in the 2017 Unit Offering, (b) 3,766,774 shares of our common stock issued in the 2017 Unit Offering, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Eric J Pearson DVM 401(k) Profit Sharing Plan FBO Eric J Pearson, (c) 4,030,187 shares of our common stock issued in the 2017 Unit Offering, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Eric J Pearson DVM 401(k) Profit Sharing Plan FBO Lianne Pearson, (d) 968,993 shares of our common stock issued in the 2017 Unit Offering, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Sunwest Trust FBO Lianne L. Pearson Roth IRA, (e) 1,234,262 shares of our common stock issued in the 2017 Unit Offering, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Sunwest Trust FBO Eric J. Pearson Roth IRA, (f) 400,000 shares of our common stock issued in the 2017 Unit Offering, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Sunwest Trust FBO Eric J. Pearson Roth IRA, (g) 7,762,809 shares of our common stock issued in the 2017 Unit Offering, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Eric J Pearson DVM 401(k) Profit Sharing Plan FBO Eric J Pearson, (h) 2,140,775 shares of our common stock issued in the 2017 Unit Offering, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Eric J Pearson DVM 401(k) Profit Sharing Plan FBO Lianne Pearson, (i) 66,596 shares of our common stock issued in the 2017(3) Unit Offering, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Sunwest Trust as Custodian for Lianne Pearson Roth IRA, (j) 3,099,921 shares of our common stock issued in the 2018 Warrant Exchange Offering, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Sunwest Trust FBO Eric J. Pearson Roth IRA, (k) 34,210 shares of our common stock issued in the 2018 Warrant Exchange Offering, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Sunwest Trust FBO Lianne L. Pearson Roth IRA, (l) 208,333 shares of our common stock issuable upon exercise of a warrant issued in the 2017 Unit Offering at an exercise price of $0.12 per share, (m) 3,766,774 shares of our common stock issuable upon exercise of a warrant issued in the 2017 Unit Offering at an exercise price of $0.12 per share, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Eric J Pearson DVM 401(k) Profit Sharing Plan FBO Eric J Pearson, (n) 4,030,187 shares of our common stock issuable upon exercise of a warrant issued in the 2017 Unit Offering at an exercise price of $0.12 per share, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Eric J Pearson DVM 401(k) Profit Sharing Plan FBO Lianne Pearson, (o) 968,993 shares of our common stock issuable upon exercise of a warrant issued in the 2017 Unit Offering at an exercise price of $0.12 per share, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Sunwest Trust FBO Lianne L. Pearson Roth IRA, (p) 1,234,262 shares of our common stock issuable upon exercise of a warrant issued in the 2017 Unit Offering at an exercise price of $0.12 per share, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Sunwest Trust FBO Eric J. Pearson Roth IRA, (q) 400,000 shares of our common stock issuable upon exercise of a warrant issued in the 2017 Unit Offering at an exercise price of $0.12 per share, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Sunwest Trust FBO Eric J. Pearson Roth IRA, (r) 7,762,809 shares of our common stock issuable upon exercise of a warrant issued in the 2017 Unit Offering at an exercise price of $0.12 per share, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Eric J Pearson DVM 401(k) Profit Sharing Plan FBO Eric J Pearson, (s) 2,140,775 shares of our common stock issuable upon exercise of a warrant issued in the 2017 Unit Offering at an exercise price of $0.12 per share, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Eric J Pearson DVM 401(k) Profit Sharing Plan FBO Lianne Pearson, and (t) 66,596 shares of our common stock issuable upon exercise of a warrant issued in the 2017(3) Unit Offering at an exercise price of $0.30 per share, which the Pearson’s may be deemed to beneficially own as the beneficiaries of the Sunwest Trust as Custodian for Lianne Pearson Roth IRA. |
| (21) | The address of Mr. James K. Schuler is 828 Fort Street Mall, Honolulu, HI 96813. Mr. Schuler does not have any position, office, contractual relationship, or other understanding with the Company regarding the management or control of the Company and accordingly, we have determined that such stockholder is not an affiliate of Cardax. |
| (22) | Represents (a) 4,673,313 shares of our common stock which Mr. Schuler may be deemed to beneficially own as the President of JKS Partners, L.P., (b) 1,960,170 shares of our common stock which Mr. Schuler may be deemed to beneficially own as the President of The Schuler Family Foundation, (c) 404,009 shares of our common stock which Mr. Schuler may be deemed to beneficially own as the Trustee of the James K. Schuler Revocable Living Trust, (d) 2,666,664 shares of our common stock issuable upon exercise of warrants issued in the 2015 Unit Offering at an exercise price of $0.10 per share, which Mr. Schuler may be deemed to beneficially own as the President of JKS Partners, L.P., (e) 1,000,000 shares of our common stock issuable upon exercise of warrants issued in the 2015 Unit Offering at an exercise price of $0.1667 per share, which Mr. Schuler may be deemed to beneficially own as the President of JKS Partners, L.P., (f) 20,396 shares of our common stock issuable upon exercise of a warrant issued in the Holdings Merger at an exercise price of $0.981 per share, which Mr. Schuler may be deemed to beneficially own as the President of JKS Partners, L.P., (g) 1,250,000 shares of our common stock issuable upon exercise of a warrant issued in the 2016-2017 Unit Offering at an exercise price of $0.08 per share, which Mr. Schuler may be deemed to beneficially own as the President of JKS Partners, L.P., (h) 1,250,000 shares of our common stock issuable upon exercise of a warrant issued in the 2016-2017 Unit Offering at an exercise price of $0.12 per share, which Mr. Schuler may be deemed to beneficially own as the President of JKS Partners, L.P., (i) 1,250,000 shares of our common stock issuable upon exercise of a warrant issued in the 2016-2017 Unit Offering at an exercise price of $0.16 per share, which Mr. Schuler may be deemed to beneficially own as the President of JKS Partners, L.P., (j) 208,333 shares of our common stock issuable upon exercise of a warrant issued in the 2017 Unit Offering at an exercise price of $0.12 per share, which Mr. Schuler may be deemed to beneficially own as the President of JKS Partners, L.P., (k) 101,983 shares of our common stock issuable upon exercise of a warrant issued in the Holdings Merger at an exercise price of $0.981 per share, which Mr. Schuler may be deemed to beneficially own as the President of The Schuler Family Foundation, (l) 11,645,962 shares of our common stock issuable upon conversion of a convertible note at an exercise price of $0.07 per share, which Mr. Schuler may be deemed to beneficially own as the President of JKS Partners, L.P., (m) 1,500,000 shares of our common stock issuable upon exercise of a warrant issued in connection with a convertible note at an exercise price of $0.07 per share, which Mr. Schuler may be deemed to beneficially own as the President of JKS Partners, L.P., (n) 3,105,590 shares of our common stock issuable upon conversion of a convertible note at an exercise price of $0.07 per share, which Mr. Schuler may be deemed to beneficially own as the President of JKS Partners, L.P., and (o) 400,000 shares of our common stock issuable upon exercise of a warrant issued in connection with a convertible note at an exercise price of $0.07 per share, which Mr. Schuler may be deemed to beneficially own as the President of JKS Partners, L.P. |
| 57 |
Certain Relationships and Related Party Transactions
On October 16, 2017, we engaged M.M. Dillon & Co., to serve as the Financial Advisor for the Company in connection with an exchange offer and related transactions. George W. Bickerstaff, III, the Chairman of our Board of Directors, is currently a Managing Director of M.M. Dillon & Co., and as such, abstained from voting on the engagement of M.M. Dillon & Co. On May 2, 2018, the terms of M.M. Dillon & Co.’s engagement were modified, and such modifications were approved by our Board of Directors (with Mr. Bickerstaff abstaining) on April 30, 2018. We raised $1.44 million in gross proceeds in the exchange offer, which closed on July 27, 2018. As the Financial Advisor in connection with the exchange offer, M.M. Dillon & Co. was paid a cash fee of 3.5% of the gross proceeds from the exchange offer, or $50,401.50, and a 5-year common stock purchase warrant with a fair market value equal to 3.5% of the gross proceeds from the exchange offer, or $50,401.50. In addition, M.M. Dillon & Co. LLC will receive compensation as our financial adviser in this offering, as described in “Underwriting - Additional Fees Incurred In Connection With This Offering”.
On June 26, 2019, we and our Chief Executive Officer, as a lender to the Company, entered into a promissory note payable in the amount of $75,000. This note accrues interest at the rate of 4.5% per annum and has been amended so that it matures on December 31, 2019.
On November 15, 2019, we and our Chief Executive Officer, as a lender to the Company, entered into a convertible note payable in the amount of $100,000. This note accrues interest payable monthly at the rate of 14% per annum and matures on June 30, 2020. This note and accrued interest thereon may convert into shares of our common stock at $0.10 per share any time at the holder’s option. If this note, or any portion thereof, has not been repaid or converted in full on or prior to the maturity date, then repayment of the unpaid principal balance plus any accrued and unpaid interest thereon, shall be amortized over the following thirty-six (36) months. We have the right to prepay this note without penalty or premium.
Our Secretary is a member of the law firm that is counsel to us in this offering and provides us with counsel in corporate and securities issues.
Other than compensation arrangements with directors and executive officers, which are described under “Executive Compensation— Employment and Consulting Agreements”, and except as described above, we have no other related-party transactions that are subject to disclosure.
Authorized Capital Stock
Our authorized share capital consists of 400,000,000 shares of our common stock, par value $0.001 per share, and 50,000,000 shares of preferred stock, par value $0.001 per share. We expect to amend our certificate of incorporation in connection with the Reverse Stock Split as described in this prospectus. The Reverse Stock Split shall not alter the number of authorized shares or par value of our common stock nor modify any voting rights or other terms of our common stock.
Common Stock
As of November 20, 2019, 137,261,594 shares of our common stock were outstanding. The outstanding shares of our common stock are validly issued, fully paid, and non-assessable.
Holders of our common stock are entitled to one vote for each share on all matters submitted to a stockholder vote. Holders of our common stock do not have cumulative voting rights. Therefore, holders of a majority of the shares of our common stock voting for the election of directors can elect all of the directors. Holders of our common stock representing a majority of the voting power of the Company’s capital stock issued, outstanding, and entitled to vote, represented in person or by proxy, are necessary to constitute a quorum at any meeting of stockholders. A vote by the holders of a majority of the Company’s outstanding shares is required to effectuate certain fundamental corporate changes such as liquidation, merger, or an amendment to the Company’s certificate of incorporation.
Holders of our common stock are entitled to share in all dividends that our Board of Directors, in its discretion, declares from legally available funds. In the event of a liquidation, dissolution or winding up, each outstanding share entitles its holder to participate pro rata in all assets that remain after payment of liabilities and after providing for each class of stock, if any, having preference over the common stock. The common stock has no pre-emptive, subscription or conversion rights and there are no redemption provisions applicable to the common stock.
Preferred Stock
As of the date of this prospectus, there were no shares of our preferred stock issued and outstanding.
Our authorized preferred stock is “blank check” preferred. Accordingly, subject to limitations prescribed by law, our Board of Directors is expressly authorized, at its discretion, to adopt resolutions to issue shares of preferred stock of any class or series, to fix the number of shares of any class or series of preferred stock and to change the number of shares constituting any series and to provide for or change the voting powers, designations, preferences and relative, participating, optional or other special rights, qualifications, limitations or restrictions thereof, including dividend rights (including whether the dividends are cumulative), dividend rates, terms of redemption (including sinking fund provisions), redemption prices, conversion rights and liquidation preferences of the shares constituting any series of the preferred stock, in each case without any further action or vote by our stockholders.
Options
We adopted our 2014 Plan, pursuant to which we may grant options or other equity incentive awards to employees or other persons on terms and conditions determined by our Board of Directors or our compensation committee. The options or other equity awards that may be granted under this plan may qualify as incentive stock options under the Internal Revenue Code of 1986, as amended. The number of shares of our common stock reserved for issuance upon the exercise or exchange of such options or other equity incentive awards accounted for 16% of our capitalization as of November 20, 2019, determined on a fully diluted basis.
| 58 |
As of the date of this prospectus, we have outstanding options to purchase an aggregate of 40,438,425 shares of our common stock under our 2014 Plan adopted and approved by our Board of Directors and our stockholders as follows:
Number of Shares Underlying Options |
Option Exercise Price | Option Expiration Date |
Option Vesting | ||||
| 19,148,909 | $ | 0.625 | February 7, 2024 | (1) | |||
| 684,984 | 0.155 | May 15, 2020 | (2) | ||||
| 4,274,606 | 0.155 | February 7, 2024 | (3) | ||||
| 1,979,246 | 0.32 | June 30, 2020 | (4) | ||||
| 2,672,830 | 0.20 | June 30, 2020 | (5) | ||||
| 713,653 | 0.49 | September 30, 2020 | (6) | ||||
| 1,091,161 | 0.27 | December 31, 2020 | (7) | ||||
| 5,075,469 | 0.06 | March 31, 2021 | (8) | ||||
| 100,000 | 0.07 | July 11, 2021 | (9) | ||||
| 27,778 | 0.15 | September 30, 2021 | (10) | ||||
| 83,333 | 0.15 | December 31, 2021 | (11) | ||||
| 78,125 | 0.185 | March 31, 2022 | (12) | ||||
| 83,333 | 0.20 | June 30, 2022 | (13) | ||||
| 400,000 | 0.50 | September 25, 2027 | (14) | ||||
| 50,000 | 0.47 | September 25, 2027 | (15) | ||||
| 1,000,000 | 0.44 | November 1, 2027 | (16) | ||||
| 41,664 | 0.37 | November 27, 2027 | (17) | ||||
| 100,000 | 0.34 | December 13, 2027 | (18) | ||||
| 500,000 | 0.16 | January 1, 2028 | (19) | ||||
| 333,334 | 0.16 | January 1, 2023 | (20) | ||||
| 1,000,000 | 0.24 | June 1, 2028 | (21) | ||||
| 1,000,000 | 0.21 | November 14, 2028 | (22) | ||||
| 40,438,425 | $ | 0.06-0.625 | 2020-2028 | ||||
| (1) | Fifty percent of these options became immediately exercisable as of February 7, 2014, and the remaining 50% vested ratably on a monthly basis through February 7, 2015. | |
| (2) | These options became immediately exercisable on February 7, 2014. | |
| (3) | These options became immediately exercisable on February 7, 2014. | |
| (4) | These options became immediately exercisable on June 30, 2015. | |
| (5) | These options became immediately exercisable on June 30, 2015. | |
| (6) | These options became immediately exercisable on September 30, 2015. | |
| (7) | These options became immediately exercisable on December 31, 2015. | |
| (8) | These options became immediately exercisable on March 31, 2016. | |
| (9) | One quarter (1/4) of the shares vested on the last day of each calendar quarter following July 11, 2016. | |
| (10) | This option became immediately exercisable on September 30, 2016. | |
| (11) | This option became immediately exercisable on December 31, 2016. | |
| (12) | This option became immediately exercisable on March 31, 2017. | |
| (13) | This option became immediately exercisable on June 30, 2017. | |
| (14) | One-fourth (1/4) of the shares vest on September 25, 2018 and one forty-eighth (1/48) of the shares vest on the last day of each full month thereafter. | |
| (15) | This option became immediately exercisable on January 31, 2018. | |
| (16) | One-fourth (1/4) of the shares vest on November 1, 2018 and one forty-eighth (1/48) of the shares vest on the last day of each full month thereafter. | |
| (17) | The shares are fully vested. | |
| (18) | One-fourth (1/4) of the shares vest on December 13, 2018 and one forty-eighth (1/48) of the shares vest on the last day of each full month thereafter. | |
| (19) | One-fourth (1/4) of the shares vest on January 1, 2019 and one forty-eighth (1/48) of the shares vest on the last day of each full month thereafter. | |
| (20) | One-twelfth (1/12) of the shares vest on the last day of each month thereafter. | |
| (21) | One twenty-fourth (1/24) of the shares vest on the last day of each month thereafter subject to certain acceleration provisions. | |
| (22) | One forty-eighth (1/48) of one-half (1/2) of the shares vest on the last day of each month thereafter, and the remaining shares vest upon certain milestones, subject to certain acceleration provisions. |
| 59 |
Each option exercise price described above is subject to adjustment for certain corporate events such as stock splits (or reverse stock splits including the Reverse Stock Split), subdivisions or combinations, reclassifications, exchanges or substitutions of our common stock or upon a merger or reorganization or similar transaction. The number of shares of our common stock underlying each option and the exercise price per share of each option stated above has not been adjusted to give effect to the Reverse Stock Split.
Warrants
As of the date of this prospectus, we have outstanding warrants to purchase an aggregate of 103,838,031 shares of our common stock as follows:
Number of Shares Underlying Warrants |
Warrant Exercise Price |
Warrant
Expiration Date | |||
| 30,000 | $ | 0.40 | November 10, 2019 | ||
| 50,000 | 0.30 | March 31, 2020 | |||
| 12,041,450 | 0.10 | March 31, 2020 | |||
| 4,515,554 | 0.1667 | March 31, 2020 | |||
| 149,000 | 0.30 | December 31, 2020 | |||
| 111,750 | 0.1667 | December 31, 2020 | |||
| 15,750,000 | 0.08 | Various dates in 2021 and 2022 | |||
| 16,250,000 | 0.12 | Various dates in 2021 and 2022 | |||
| 16,250,000 | 0.16 | Various dates in 2021 and 2022 | |||
| 31,453,788 | 0.12 | Various dates in 2022 | |||
| 416,595 | 0.30 | Various dates in 2022 | |||
| 295,747 | 0.981 | Various dates in 2019 | |||
| 159,058 | 0.981 | Various dates in 2020 | |||
| 177,164 | 0.25 | December 31, 2019 | |||
| 558,750 | 0.08 | May 3, 2022 | |||
| 558,750 | 0.12 | May 3, 2022 | |||
| 558,750 | 0.16 | May 3, 2022 | |||
| 315,010 | 0.21 | July 27, 2023 | |||
| 816,665 | 0.20 | Various dates in 2024 | |||
| 500,000 | 0.20 | April 18, 2024 | |||
| 1,500,000 | 0.07 | * | July 19, 2024 | ||
| 200,000 | 0.12 | September 4, 2024 | |||
| 100,000 | 0.12 | September 25, 2024 | |||
| 50,000 | 0.12 | October 3, 2024 | |||
| 50,000 | 0.12 | October 10, 2024 | |||
| 400,000 | 0.07 | * | October 16, 2024 | ||
| 250,000 | 0.15 | October 23, 2024 | |||
| 250,000 | 0.20 | October 23, 2024 | |||
| 50,000 | 0.12 | October 29, 2024 | |||
30,000 |
0.07 |
November 8, 2024 | |||
| 103,838,031 | $ | 0.07-0.981 | 2019-2024 | ||
Each warrant exercise price described above is subject to adjustment for certain corporate events such as stock splits (or reverse stock splits including the Reverse Stock Split), subdivisions or combinations, reclassifications, exchanges or substitutions of our common stock or upon a merger or reorganization or similar transaction. In addition, each warrant exercise price notated above with a “*” is subject to adjustment upon the issuance of our common stock or securities convertible into our common stock at a price per share less than the then prevailing exercise price, other than specified exempt issuances. The number of shares of our common stock underlying each warrant and the exercise price per share of each warrant stated above has not been adjusted to give effect to the Reverse Stock Split. The above description of warrants is qualified in its entirety by reference to the forms of such warrants filed as exhibits to the registration statement of which this prospectus forms a part.
| 60 |
Convertible Promissory Notes
We entered into convertible notes payable with lenders as set forth in the table below. Each of these notes and accrued interest thereon may convert into shares of our common stock at the conversion price set forth in the table below. We have the right to prepay each of these notes without penalty or premium.
Certain of these notes were issued with detachable five-year warrants to purchase shares of our common stock as set forth in the table below and described above under “Description of Securities – Warrants”.
Issuance Date | Principal Amount | Original Issue Discount | Gross Proceeds | Interest Rate | Maturity Date | Note Conversion Price Per Share | Number of Shares Underlying Warrants | Warrant Exercise Price Per Share | ||||||||||||||||||||||||
| April 18, 2019 | $ | 150,000 | $ | - | $ | 150,000 | 10 | %(1) | December 31, 2019 | $ | 0.07 | (4,5) | 500,000 | $ | 0.20 | |||||||||||||||||
| July 19, 2019 | 815,217 | 65,217 | 750,000 | 8 | %(2) | June 30, 2020 | 0.07 | (4,6) | 1,500,000 | 0.07 | (8) | |||||||||||||||||||||
| September 4, 2019 | 108,696 | 8,696 | 100,000 | 8 | %(2) | June 30, 2020 | (3) | 0.12 | (7) | 200,000 | 0.12 | |||||||||||||||||||||
| September 25, 2019 | 54,348 | 4,348 | 50,000 | 8 | %(2) | June 30, 2020 | (3) | 0.12 | (7) | 100,000 | 0.12 | |||||||||||||||||||||
| October 3, 2019 | 27,174 | 2,174 | 25,000 | 8 | %(2) | June 30, 2020 | (3) | 0.12 | (7) | 50,000 | 0.12 | |||||||||||||||||||||
| October 10, 2019 | 27,174 | 2,174 | 25,000 | 8 | %(2) | June 30, 2020 | (3) | 0.12 | (7) | 50,000 | 0.12 | |||||||||||||||||||||
| October 16, 2019 | 217,391 | 17,391 | 200,000 | 8 | %(2) | June 30, 2020 | 0.07 | (4,6) | 400,000 | 0.07 | (8) | |||||||||||||||||||||
| October 23, 2019 | 108,696 | 8,696 | 100,000 | 8 | %(2) | June 30, 2020 | (3) | 0.12 | (7) | 250,000 250,000 | 0.15 0.20 | |||||||||||||||||||||
| October 29, 2019 | 27,174 | 2,174 | 25,000 | 8 | %(2) | June 30, 2020 | (3) | 0.12 | (7) | 50,000 | 0.12 | |||||||||||||||||||||
| November 8, 2019 | 16,304 | 1,304 | 15,000 | 8 | %(2) | June 30, 2020 | (3) | 0.07 | (7) | 30,000 | 0.07 | |||||||||||||||||||||
| November 15, 2019 | 100,000 | - | 100,000 | 14 | %(2) | June 30, 2020 | (3) | 0.10 | (7) | - | - | |||||||||||||||||||||
| Total | $ | 1,652,174 | $ | 112,174 | $ | 1,540,000 | 8-14 | % | 2019-2020 | $ | 0.07-0.12 | 3,380,000 | $ | 0.07-0.20 | ||||||||||||||||||
| (1) | Accrued interest on this note is payable upon maturity. | |
| (2) | Accrued interest on this note is payable monthly in cash. | |
| (3) | If this note, or any portion thereof, has not been repaid or converted in full on or prior to the maturity date, then repayment of the unpaid principal balance plus any accrued and unpaid interest thereon, shall be amortized over the following thirty-six (36) months. | |
| (4) | The conversion price of this note is subject to adjustment upon the issuance of our common stock or securities convertible into our common stock at a price per share less than the then prevailing conversion price, other than specified exempt issuances. | |
| (5) | This note and accrued interest thereon may convert into shares of our common stock any time at the holder’s option or automatically upon maturity provided the 20-day volume weighted average price per share of our common stock upon maturity is at least $0.12 per share. | |
| (6) | This note and accrued interest thereon may convert into shares of our common stock any time at the holder’s option or automatically upon a qualified financing of at least $5 million at the lower of the conversion price then in effect or a 25% discount to the offering price. | |
| (7) | This note and accrued interest thereon may convert into shares of our common stock any time at the holder’s option. | |
| (8) | The exercise price of this warrant shall be adjusted in accordance with any adjustment to the conversion price of this note. |
The above description of convertible promissory notes and related warrants is qualified in its entirety by reference to the forms of such securities filed as exhibits to the registration statement of which this prospectus forms a part.
Other Convertible Securities
Other than as described above, we do not have outstanding any options, warrants, or other securities that are convertible into, or exchangeable for, shares of our common stock.
Registration Rights
Between 2015 and 2017, we sold approximately $7,000,000 of subscriptions in securities under separate subscription agreements (each, a “Subscription Agreement”), by and between the Company and investors, pursuant to which we issued and sold to the investors units, consisting of shares of our common stock and warrants to purchase shares of our common stock (“Units”). Under the terms of the Subscription Agreements, the Company agreed to register such Units, which were registered under a previous registration statement.
| 61 |
Anti-Takeover Provisions
Amended and Restated Certificate of Incorporation and Bylaws
Our Amended and Restated Bylaws provides that our stockholders do not have cumulative voting rights, and thus stockholders holding a majority of the voting power of our shares of our common stock outstanding will be able to elect all of our directors. The authorized number of directors may be changed only by resolution of our Board of Directors, and vacancies and newly created directorships on our Board of Directors may, except as otherwise required by law or determined by our board, only be filled by a majority vote of the directors then serving on our Board of Directors, even though less than a quorum (except, that (i) stockholders removing any director may at the same meeting fill the vacancy and (ii) if the directors fail to fill any such vacancy, the stockholders may at any special meeting called for that purpose fill such vacancy.). A special meeting of stockholders may be called only by our Board of Directors, the Chairman of our Board of Directors, or by our Chief Executive Officer. Our amended and restated bylaws also provide that stockholders seeking to present proposals before a meeting of stockholders to nominate candidates for election as directors at a meeting of stockholders must provide timely advance notice in writing, and specify requirements as to the form and content of a stockholder’s notice.
The foregoing provisions make it more difficult for our existing stockholders to replace our Board of Directors as well as for another party to obtain control of our company by replacing our Board of Directors. Since our Board of Directors has the power to retain and discharge our officers, these provisions could also make it more difficult for existing stockholders or another party to effect a change in management. In addition, the authorization of undesignated preferred stock makes it possible for our Board of Directors to issue preferred stock with voting or other rights or preferences that could impede the success of any attempt to change the control of our company. These provisions are intended to enhance the likelihood of continued stability in the composition of our Board of Directors and its policies and to discourage certain types of transactions that may involve an actual or threatened acquisition of our company. These provisions are also designed to reduce our vulnerability to an unsolicited acquisition proposal and to discourage certain tactics that may be used in proxy fights. However, such provisions could have the effect of discouraging others from making tender offers for our shares and may have the effect of deterring hostile takeovers or delaying changes in control of our company or our management. As a consequence, these provisions also may inhibit fluctuations in the market price of our stock that could result from actual or rumored takeover attempts.
In addition, our authorized but unissued common shares could be used by our Board of Directors for defensive purposes against a hostile takeover attempt, including (by way of example) the private placement of shares or the granting of options to purchase shares to persons or entities sympathetic to, or contractually bound to support, management. We have no such present arrangement or understanding with any person. Further, our common stock may be reserved for issuance upon exercise of stock purchase rights designed to deter hostile takeovers, commonly known as a “poison pill.”
Section 203 of the Delaware General Corporation Law
We are subject to Section 203 of the Delaware General Corporation Law, or Section 203, which prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years after the date that such stockholder became an interested stockholder, with the following exceptions:
| ● | before such date, our Board of Directors approved either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder; | |
| ● | upon closing of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least eighty-five percent (85%) of the voting stock of the corporation outstanding at the time the transaction began, excluding for purposes of determining the voting stock outstanding (but not the outstanding voting stock owned by the interested stockholder) those shares owned by: (i) persons who are directors and also officers; and (ii) employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or | |
| ● | on or after such date, the business combination is approved by our Board of Directors and authorized at an annual or special meeting of the stockholders, and not by written consent, by the affirmative vote of at least 66 2∕3% of the outstanding voting stock that is not owned by the interested stockholder. |
In general, Section 203 defines business combination to include the following:
| ● | any merger or consolidation involving the corporation and the interested stockholder; | |
| ● | any sale, transfer, pledge, or other disposition of ten percent (10%) or more of the assets of the corporation involving the interested stockholder; | |
| ● | subject to certain exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; | |
| ● | any transaction involving the corporation that has the effect of increasing the proportionate share of the stock or any class or series of the corporation beneficially owned by the interested stockholder; or | |
| ● | the receipt by the interested stockholder of the benefit of any loss, advances, guarantees, pledges, or other financial benefits by or through the corporation. |
In general, Section 203 defines an “interested stockholder” as an entity or person who, together with the person’s affiliates and associates, beneficially owns, or within three years prior to the time of determination of interested stockholder status did own, fifteen percent (15%) or more of the outstanding voting stock of the corporation.
Limitation on Liability and Indemnification Matters
See the section of this prospectus entitled “Management — Indemnification”.
Listing
Our common stock is traded on the OTCQB under the trading symbol “CDXI”.
Transfer Agent
Our independent transfer agent is VStock Transfer, LLC. VStock Transfer’s address is 18 Lafayette Place, Woodmere, NY 11598.
| 62 |
DESCRIPTION OF THE SECURITIES WE ARE OFFERING
We are offering ______ units, each unit consisting of ______ shares of our common stock together with ______ Purchase Warrants to purchase up to an aggregate of ______ shares of our common stock. The shares of our common stock and related Purchase Warrants will be issued separately. We are also registering the shares of our common stock issuable upon exercise of the Purchase Warrants offered hereby.
Common Stock
The material terms and provisions of our common stock are described under the caption “Description of Securities”.
Purchase Warrants to be Issued in this Offering
The following summary of certain terms and provisions of the Purchase Warrants that are being offered hereby is not complete and is subject to, and qualified in its entirety by the provisions of, the Purchase Warrant. Prospective investors should carefully review the terms and provisions of the form of Purchase Warrant for a complete description of the terms and conditions of the Purchase Warrants.
Exercisability. The Purchase Warrants are exercisable beginning on the date of original issuance and at any time up to the date that is five years after their original issuance. The Purchase Warrants will be exercisable, at the option of each holder, in whole or in part by delivering to us a duly executed exercise notice and, at any time a registration statement registering the issuance of the shares of our common stock underlying the Purchase Warrants under the Securities Act is effective and available for the issuance of such shares, or an exemption from registration under the Securities Act is available for the issuance of such shares, by payment in full in immediately available funds for the number of shares of our common stock purchased upon such exercise. If a registration statement registering the issuance of the shares of our common stock underlying the Purchase Warrants under the Securities Act is not effective or available and an exemption from registration under the Securities Act is not available for the issuance of such shares, the holder may, in its sole discretion, elect to exercise the Purchase Warrant through a cashless exercise, in which case the holder would receive upon such exercise the net number of shares of our common stock determined according to the formula set forth in the Purchase Warrant. No fractional shares of our common stock will be issued in connection with the exercise of a Purchase Warrant. In lieu of fractional shares, we will round up to the next whole share.
Exercise Limitation. A holder will not have the right to exercise any portion of the Purchase Warrant if the holder (together with its affiliates) would beneficially own in excess of 4.99% (or, upon election of the holder, 9.99%) of the number of shares of our common stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the Purchase Warrants. However, any holder may increase or decrease such percentage, provided that any increase will not be effective until the 61st day after such election. This limitation is referred to as the “Beneficial Ownership Limitation”.
Exercise Price. The Purchase Warrants will have an exercise price of $______ per share. The exercise price is subject to appropriate adjustment in the event of certain stock dividends and distributions, stock splits, stock combinations, reclassifications, or similar events affecting our common stock and also upon any distributions of assets, including cash, stock, or other property to our stockholders.
Transferability. Subject to applicable laws, the Purchase Warrants may be offered for sale, sold, transferred, or assigned without our consent.
Exchange Listing. There is no established public trading market for the Purchase Warrants. We intend to seek a listing for the Purchase Warrants on Nasdaq under the symbol “CDXIW,” however we cannot assure you that we will be successful listing the Purchase Warrants on Nasdaq or, if successful, that an active trading market for the Purchase Warrants will develop or be sustained.
Fundamental Transactions. If a fundamental transaction as defined in the Purchase Warrant occurs, then the successor entity will succeed to, and be substituted for us, and may exercise every right and power that we may exercise and will assume all of our obligations under the Purchase Warrants with the same effect as if such successor entity had been named in the Purchase Warrant itself. If holders of our common stock are given a choice as to the securities, cash or property to be received in a fundamental transaction, then the holder shall be given the same choice as to the consideration it receives upon any exercise of the Purchase Warrant following such fundamental transaction.
Rights as a Stockholder. Except as otherwise provided in the Purchase Warrants or by virtue of such holder’s ownership of shares of our common stock, the holder of a Purchase Warrant does not have the rights or privileges of a holder of our common stock, including any voting rights, until the holder exercises the Purchase Warrant. The Purchase Warrants provide that if we declare or make any dividend or other distribution of our assets (or rights to acquire its assets) to holders of shares of our common stock, by way of return of capital or otherwise (including, without limitation, any distribution of cash, stock or other securities, property or options by way of a dividend, spin off, reclassification, corporate rearrangement, scheme of arrangement or other similar transaction) (a “Distribution”), at any time after the issuance of the Purchase Warrant, then, in each such case, the holder of the Purchase Warrant shall be entitled to participate in such Distribution to the same extent that the holder of the Purchase Warrant would have participated therein if the holder of the Purchase Warrant had held the number of shares of common stock acquirable upon complete exercise of this Warrant (without regard to any limitations on exercise of such Purchase Warrant, including without limitation, the Beneficial Ownership Limitation) immediately before the date of which a record is taken for such Distribution, or, if no such record is taken, the date as of which the record holder of the Purchase Warrants of shares of common stock are to be determined for the participation in such Distribution (provided, however, to the extent that the holder of the Purchase Warrant’s right to participate in any such Distribution would result in the holder of the Purchase Warrant exceeding the Beneficial Ownership Limitation, then the holder of the Purchase Warrant shall not be entitled to participate in such Distribution to such extent (or in the beneficial ownership of any shares of common stocks a result of such Distribution to such extent) and the portion of such Distribution shall be held in abeyance for the benefit of the holder of the Purchase Warrant until such time, if ever, as its right thereto would not result in the holder of the Purchase Warrant exceeding the Beneficial Ownership Limitation).
| 63 |
SHARES ELIGIBLE FOR FUTURE SALE
Prior to this offering, a very limited public market exists for our common stock. Future sales of our common stock in the public market, or the availability of such shares for sale in the public market, could adversely affect market prices prevailing from time to time. As described below, approximately ______% of our common stock outstanding immediately after this offering will be subject to contractual and legal restrictions on resale. Nevertheless, sales of our common stock eligible for sale in the public market immediately after this offering, or sales of other shares eligible for sale after such restrictions lapse, or the perception that those sales may occur, could adversely affect the prevailing market price at such time and our ability to raise equity capital in the future.
Based on the number of shares outstanding as of ______, upon the closing of this offering and assuming no exercise of the underwriters’ option to purchase additional shares and/or warrants, ______ shares of our common stock will be outstanding, assuming no outstanding options or warrants are exercised. All of the shares of our common stock sold in this offering will be freely tradable without restrictions or further registration under the Securities Act, except for any shares sold to our “affiliates,” as that term is defined under Rule 144 under the Securities Act. Of the remaining ______ shares of our common stock held by existing stockholders, ______ shares are “restricted securities,” as that term is defined in Rule 144 under the Securities Act. Restricted securities may be sold in the public market only if registered or if their resale qualifies for exemption from registration described pursuant to Rule 144 promulgated under the Securities Act.
As a result of the lock-up agreements referred to below and the provisions of Rule 144 under the Securities Act, the shares of our common stock (excluding the shares sold in this offering) that will be available for sale in the public market are as follows:
| Number of Shares and % of Total Outstanding | Date Available for Sale into Public Market | |
| ______ shares, or ______% | On the date of this prospectus. | |
| ______ shares, or ______% | Six (6) months after the date of this prospectus, due to lock-up agreements between the holders of these shares and the underwriters. However, the representatives of the underwriters, acting together, can waive the provisions of these lock-up agreements and allow these stockholders to sell their shares at any time. | |
| ______ shares, or ______% | From ______ to ______, due to the restrictions subject to Rule 144. |
| 64 |
Maxim Group LLC is acting as sole bookrunner and as representative of the underwriters (the “Representative”). Subject to the terms and conditions of an underwriting agreement between us and the Representative, we have agreed to sell to each underwriter named below, and each underwriter named below has severally agreed to purchase, at the public offering price less the underwriting discounts set forth on the cover page of this prospectus, the number of shares of our common stock and corresponding Purchase Warrants listed next to its name in the following table:
| Name of Underwriter | Number of Shares | Number of Purchase Warrants | ||||||
| Maxim Group LLC | ||||||||
| Total | ||||||||
The underwriters are committed to purchase all the shares of our common stock and corresponding Purchase Warrants offered by this prospectus if they purchase any shares of our common stock and corresponding Purchase Warrants. The underwriting agreement also provides that if an underwriter defaults, the purchase commitments of non-defaulting underwriters may be increased or the offering may be terminated. The underwriters are not obligated to purchase the shares of our common stock and/or corresponding Purchase Warrants covered by the underwriters’ over-allotment option described below. The underwriters are offering the shares of our common stock and corresponding Purchase Warrants, subject to prior sale, when, as and if issued to and accepted by them, subject to approval of legal matters by their counsel, and other conditions contained in the underwriting agreement, such as the receipt by the underwriters of officer’s certificates and legal opinions. The underwriters reserve the right to withdraw, cancel, or modify offers to the public and to reject orders in whole or in part.
Over-Allotment Option
We have granted to the underwriters an option, exercisable no later than 45 calendar days after the date of the underwriting agreement, to purchase up to an additional ______ shares of our common stock and/or warrants to purchase ______ shares of our common stock at the public offering price listed on the cover page of this prospectus, less underwriting discounts. The underwriters may exercise this option only to cover over-allotments, if any, made in connection with this offering and may exercise this option to purchase additional shares and/or Purchase Warrants. To the extent the option is exercised and the conditions of the underwriting agreement are satisfied, we will be obligated to sell to the underwriters, and the underwriters will be obligated to purchase, these additional shares of our common stock and/or Purchase Warrants.
Discounts and Commissions
We have agreed to provide the underwriters with an underwriting discount equal to nine percent (9.0%) of the public offering price; provided that the underwriting discount shall be equal to four and one-half percent (4.5%) of the public offering price for any securities sold in this offering to any of our current stockholders.
The Representative has advised us that the underwriters propose to offer the shares and corresponding Purchase Warrants directly to the public at the public offering price set forth on the cover of this prospectus. In addition, the Representative may offer some of the shares and corresponding Purchase Warrants to other securities dealers at such price less a concession of up to $ ______ per share of our common stock and related Purchase Warrant. After the offering to the public, the offering price and other selling terms may be changed by the Representative without changing the Company’s proceeds from the underwriters’ purchase of the shares and corresponding Purchase Warrants.
The following table summarizes the public offering price, underwriting discounts, and proceeds before estimated offering fees and expenses to us assuming both no exercise and full exercise of the underwriters’ option to purchase additional shares of our common stock and/or Purchase Warrants. The underwriting discounts are equal to the public offering price per share and related Purchase Warrants less the amount per share the underwriters pay us for the shares of our common stock and Purchase Warrants.
| Per Unit | Total (No Exercise) |
Total (Full Exercise) |
||||||||||
| Public offering price | $ | $ | $ | |||||||||
| Underwriting discounts(1) | ||||||||||||
| Proceeds to us, before fees and expenses(2) | $ | $ | $ | |||||||||
| (1) | Assumes an underwriting discount of 9.0% of the public offering price for all securities sold in this offering. | |
| (2) | We estimate that the total expenses of the offering, including registration, filing and listing fees, printing fees and legal and accounting expenses will be approximately $385,000, all of which are payable by us. This figure includes expense reimbursements we have agreed to pay Maxim for reimbursement of its expenses related to the offering up to a maximum aggregate expense allowance of $100,000 (including any advance toward the maximum expense allowance). For a description of other fees payable by us, please refer to the “Financial Advisory Fees” section below. |
| 65 |
Determination of Offering Price
The offering price has been negotiated between the representatives of the Underwriter and us. In determining the offering of the common stock, the following factors were considered:
| ● | prevailing market conditions; | |
| ● | our historical performance and capital structure; | |
| ● | estimates of our business potential and earnings prospects; | |
| ● | an overall assessment of our management; and | |
| ● | the consideration of these factors in relation to market valuation of companies in related businesses. |
Representative’s Warrants
At the Closing, the Company shall grant to the Representative (or its designated affiliates) share purchase warrants (the “Representative’s Warrants”) covering a number of shares of common stock equal to four and one-half percent (4.5%) of the total number of shares of common stock being sold in this Offering. The Representative’s Warrants will be non-exercisable for six (6) months after the effective date of the registration statement of which this prospectus forms a part and will expire five (5) years after such effective date. The Representative’s Warrants will be exercisable at a price equal to 125.0% of the public offering price in connection with the Offering. The Representative’s Warrants shall not be redeemable. The Company will register the shares of Common Stock underlying the Representative’s Warrants under the Act and will file all necessary undertakings in connection therewith. The Representative’s Warrants may not be sold, transferred, assigned, pledged, or hypothecated, or be the subject of any hedging, short sale, derivative, put, or call transaction that would result in the effective economic disposition of the securities by any person for a period of 180 days following the effective date of the registration statement of which this prospectus forms a part, except that they may be assigned, in whole or in part, to any officer or partner of the Representative and to members of the underwriting syndicate or selling group and officers and partners thereof. The Representative’s Warrants may be exercised as to all or a lesser number of shares of common stock for a period of five (5) years after the effective date of the registration statement of which this prospectus forms a part, will provide for cashless exercise and will contain provisions for one demand registration of the sale of the underlying shares of common stock at the Company’s expense, an additional demand registration at the warrant holders’ expense, and unlimited “piggyback” registration rights at the Company’s expense. The Representative’s Warrants shall further provide for anti-dilution protection (adjustment in the number and price of such warrants and the shares underlying such warrants) resulting from corporate events (which would include dividends, reorganizations, mergers, etc.) when the public shareholders have been proportionally affected and otherwise in compliance with FINRA Rule 5110(f)(2)(G)(vi).
Lock-Up Agreements
We and each of our officers, directors, and certain holders of our common stock as of the date of this prospectus have agreed, subject to certain exceptions, not to offer, issue, sell, contract to sell, encumber, grant any option for the sale of, or otherwise dispose of any shares of our common stock or other securities convertible into or exercisable or exchangeable for shares of our common stock for a period of six (6) months after this offering is completed without the prior written consent of Maxim Group LLC.
Maxim Group LLC may in its sole discretion and at any time without notice release some or all of the shares subject to lock-up agreements prior to the expiration of the lock-up period. When determining whether or not to release shares from the lock-up agreements, the representative will consider, among other factors, the security holder’s reasons for requesting the release, the number of shares for which the release is being requested and market conditions at the time.
Right of First Refusal
We have granted the Representative a right of first refusal, for a period of eighteen (18) months from the commencement of sales of this offering, to act as (a) lead managing underwriter and book runner or sole placement agent for any and all future public and private equity and debt offerings (each, a “Subsequent Offering”), and/or (b) to act as the Company’s exclusive advisor with respect to any merger, acquisition, sale of stock or assets (in which the Company may be the acquiring or acquired entity), joint venture, strategic alliance, or other similar transaction (each, a “Transaction”) during such eighteen (18) month period, of the Company, or any successor to or subsidiary of the Company.
Indemnification
We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act, and to contribute to payments that the underwriters may be required to make for these liabilities.
| 66 |
Price Stabilization, Short Positions, and Penalty Bids
In connection with this offering, the underwriters may engage in transactions that stabilize, maintain or otherwise affect the price of our common stock. Specifically, the underwriters may over-allot in connection with this offering by selling more shares than are set forth on the cover page of this prospectus. This creates a short position in our common stock for its own account. The short position may be either a covered short position or a naked short position. In a covered short position, the number of shares common stock over-allotted by the underwriters is not greater than the number of shares of our common stock that they may purchase in the over-allotment option. In a naked short position, the number of shares of our common stock involved is greater than the number of shares common stock in the over-allotment option. To close out a short position, the underwriters may elect to exercise all or part of the over-allotment option. The underwriters may also elect to stabilize the price of our common stock or reduce any short position by bidding for, and purchasing, common stock in the open market.
The underwriters may also impose a penalty bid. This occurs when a particular underwriter or dealer repays selling concessions allowed to it for distributing a security in this offering because the underwriter repurchases that security in stabilizing or short covering transactions.
Finally, the underwriters may bid for, and purchase, shares of our common stock in market making transactions, including “passive” market making transactions as described below.
These activities may stabilize or maintain the market price of our common stock at a price that is higher than the price that might otherwise exist in the absence of these activities. The underwriters are not required to engage in these activities, and may discontinue any of these activities at any time without notice.
In connection with this offering, the underwriters and selling group members, if any, or their affiliates may engage in passive market making transactions in our common stock immediately prior to the commencement of sales in this offering, in accordance with Rule 103 of Regulation M under the Exchange Act. Rule 103 generally provides that:
| ● | a passive market maker may not effect transactions or display bids for our common stock in excess of the highest independent bid price by persons who are not passive market makers; | |
| ● | net purchases by a passive market maker on each day are generally limited to 30% of the passive market maker’s average daily trading volume in our common stock during a specified two-month prior period or 200 shares, whichever is greater, and must be discontinued when that limit is reached; and | |
| ● | passive market making bids must be identified as such. |
Electronic Distribution
A prospectus in electronic format may be made available on a website maintained by the representatives of the underwriters and may also be made available on a website maintained by other underwriters. The underwriters may agree to allocate a number of shares to underwriters for sale to their online brokerage account holders. Internet distributions will be allocated by the representatives of the underwriters to underwriters that may make Internet distributions on the same basis as other allocations. In connection with the offering, the underwriters or syndicate members may distribute prospectuses electronically. No forms of electronic prospectus other than prospectuses that are printable as Adobe® PDF will be used in connection with this offering.
The underwriters have informed us that they do not expect to confirm sales of shares offered by this prospectus to accounts over which they exercise discretionary authority.
Other than the prospectus in electronic format, the information on any underwriter’s website and any information contained in any other website maintained by an underwriter is not part of the prospectus or the registration statement of which this prospectus forms a part, has not been approved and/or endorsed by us or any underwriter in its capacity as underwriter and should not be relied upon by investors.
| 67 |
Certain Relationships
Certain of the underwriters and their affiliates may provide, from time to time, investment banking and financial advisory services to us in the ordinary course of business, for which they may receive customary fees and commissions.
Financial Advisory Fees
M.M. Dillon & Co. LLC and Primary Capital, LLC are broker dealers and FINRA member firms that have provided us with certain financial advisory services in connection with our prior issuance of securities. Our agreements with each of these firms were entered into in connection with services provided for the private placement of our securities unrelated to this offering. Each agreement provides for compensation in any such private placement including a tail fee for investments by a person that was introduced to us by the respective broker dealer in the event the investor also purchases our securities in any subsequent offering.
M.M. Dillon & Co. LLC will continue to provide us with financial advisory and consulting services, however, M.M. Dillion is neither engaged in, nor affiliated with any entity that is engaged in, the solicitation or distribution of this offering. Likewise, Primary Capital, LLC is not participating in this offering within the meaning of FINRA Rule 5110(a)(5).
On the date that this registration statement that includes this prospectus is effective, our agreements with each of M.M. Dillon & Co. LLC and Primary Capital, LLC will be terminated, we will have no obligations to either firm other than for certain provisions that survive termination, and will pay these firms termination fees as follows:
| ● | We will pay M.M. Dillon & Co. LLC a cash fee equal to one percent (1%) of the gross proceeds of this offering and an equity fee of warrants with the same terms and conditions as the Representative Warrants covering a number of shares of common stock equal to one percent (1%) of the total number of shares of common stock being sold in this offering; and | |
| ● | We will pay Primary Capital, LLC a cash fee equal to one percent (1%) of the gross proceeds of this offering. |
The validity of the shares of our common stock covered by this prospectus will be passed upon by Allegaert Berger & Vogel LLP, New York, New York.
The financial statements included in this prospectus have been so included in reliance on the report of KBL, LLP, an independent registered public accounting firm, given on the authority of said firm as experts in auditing and accounting.
Where You Can Find Additional Information
We have filed with the SEC under the Securities Act a registration statement on Form S-1 relating to the shares of our common stock that may be sold in this offering. The registration statement, including the attached exhibits and schedules, contains additional relevant information about us and our capital stock. This prospectus does not contain all of the information set forth in the registration statement and the exhibits and schedules thereto. For further information about us and our common stock, you should refer to the registration statement, including the exhibits and schedules thereto. Statements contained in this prospectus as to the contents of any contract or other document referred to are not necessarily complete and in each instance, if such contract or document is filed as an exhibit, reference is made to the copy of such contract or other document filed as an exhibit to the registration statement, each statement being qualified in all respects by such reference. You may inspect a copy of the registration statement and the exhibits and schedules thereto without charge at the Public Reference Room of the SEC at 100 F Street, N.E., Washington, D.C. 20549. You may obtain copies of all or any part of the registration statement from such office at prescribed rates. You may also obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. In addition, the SEC maintains an Internet website, which is located at http://www.sec.gov, that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. You may access the registration statement, of which this prospectus is a part, at the SEC’s Internet website. In addition to disclosing current information pursuant to Section 13 or 15(d) of the Exchange Act and for reports of information required to be disclosed by Regulation FD through our SEC filings, we also intend to disclose such current information through our investor relations website, press releases, and public conference calls.
| 68 |
Cardax, Inc., and Subsidiary
December 31, 2018 and 2017
Contents
| Page | |
| CONSOLIDATED FINANCIAL STATEMENTS: | |
| Report of Independent Registered Public Accounting Firm | F-2 |
| Consolidated balance sheets | F-3 |
| Consolidated statements of operations | F-4 |
| Consolidated statement of changes in stockholders’ deficit | F-5 |
| Consolidated statements of cash flows | F-6 |
| Notes to the consolidated financial statements | F-7 |
Cardax, Inc., and Subsidiary
September 30, 2019 and 2018
Contents
| Page | |
| CONDENSED CONSOLIDATED FINANCIAL STATEMENTS: | |
| Condensed consolidated balance sheets | F-27 |
| Condensed consolidated statements of operations | F-28 |
| Condensed consolidated statement of changes in stockholders’ deficit | F-29 |
| Condensed consolidated statements of cash flows | F-31 |
| Notes to the condensed consolidated financial statements | F-32 |
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
of Cardax, Inc. and Subsidiaries
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Cardax, Inc. and Subsidiaries (the “Company”) as of December 31, 2018 and 2017, the related consolidated statements of operations, changes in stockholders’ deficit, and cash flows for the years then ended, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the consolidated financial position of the Company as of December 31, 2018 and 2017, and the results of its consolidated operations and its cash flows for the years then ended in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal controls over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Going Concern Consideration
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has sustained significant operating losses and needs to obtain additional financing to continue the services they provide. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ KBL, LLP
We have served as the Company’s auditor since 2013.
KBL, LLP
New York, NY
March 27, 2019
| F-2 |
CONSOLIDATED BALANCE SHEETS
As of December 31,
| 2018 | 2017 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS | ||||||||
| Cash | $ | 243,753 | $ | 2,236,837 | ||||
| Accounts receivable | 157,082 | 37,243 | ||||||
| Inventories | 1,480,380 | 340,425 | ||||||
| Deposits and other assets | 119,066 | 90,831 | ||||||
| Prepaid expenses | 24,083 | 22,838 | ||||||
| Total current assets | 2,024,364 | 2,728,174 | ||||||
| PROPERTY AND EQUIPMENT, net | - | 1,901 | ||||||
| INTANGIBLE ASSETS, net | 434,534 | 426,610 | ||||||
| TOTAL ASSETS | $ | 2,458,898 | $ | 3,156,685 | ||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT | ||||||||
| CURRENT LIABILITIES | ||||||||
| Accrued payroll and payroll related expenses | $ | 3,428,011 | $ | 3,404,610 | ||||
| Accounts payable and accrued expenses | 1,996,097 | 603,391 | ||||||
| Fees payable to directors | 418,546 | 418,546 | ||||||
| Accrued separation costs, current portion | 9,000 | - | ||||||
| Employee settlement | 50,000 | 50,000 | ||||||
| Total current liabilities | 5,901,654 | 4,476,547 | ||||||
| ACCRUED SEPERATION COSTS, net of current portion | 92,635 | 85,615 | ||||||
| COMMITMENTS AND CONTINGENCIES | - | - | ||||||
| Total liabilities | 5,994,289 | 4,562,162 | ||||||
| STOCKHOLDERS’ DEFICIT | ||||||||
| Preferred Stock - $0.001 par value; 50,000,000 shares authorized, 0 shares issued and outstanding as of December 31, 2018 and 2017, respectively | - | - | ||||||
| Common stock - $0.001 par value; 400,000,000 shares authorized, 133,888,573 and 122,674,516 shares issued and outstanding as of December 31, 2018 and 2017, respectively | 133,889 | 122,675 | ||||||
| Additional paid-in-capital | 58,274,038 | 56,401,069 | ||||||
| Deferred compensation | - | (10,125 | ) | |||||
| Accumulated deficit | (61,943,318 | ) | (57,919,096 | ) | ||||
| Total stockholders’ deficit | (3,535,391 | ) | (1,405,477 | ) | ||||
| TOTAL LIABILITIES AND STOCKHOLDERS’ DEFICIT | $ | 2,458,898 | $ | 3,156,685 | ||||
The accompanying notes are an integral part of these Consolidated Financial Statements.
| F-3 |
CONSOLIDATED STATEMENTS OF OPERATIONS
For the years ended December 31,
| 2018 | 2017 | |||||||
| REVENUES, net | $ | 1,510,875 | $ | 610,323 | ||||
| COST OF GOODS SOLD | 699,852 | 274,707 | ||||||
| GROSS PROFIT | 811,023 | 335,616 | ||||||
| OPERATING EXPENSES: | ||||||||
| Salaries and wages | 1,591,949 | 830,922 | ||||||
| Selling, general, and administrative expenses | 1,493,819 | 702,168 | ||||||
| Professional fees | 797,833 | 435,749 | ||||||
| Stock based compensation | 650,271 | 242,146 | ||||||
| Research and development | 269,077 | 97,479 | ||||||
| Depreciation and amortization | 30,569 | 29,422 | ||||||
| Total operating expenses | 4,833,518 | 2,337,886 | ||||||
| Loss from operations | (4,022,495 | ) | (2,002,270 | ) | ||||
| OTHER INCOME (EXPENSE): | ||||||||
| Other income | 556 | 17,253 | ||||||
| Interest income | 1,944 | 3,320 | ||||||
| Interest expense | (4,227 | ) | (3,537 | ) | ||||
| Total other (expense) income, net | (1,727 | ) | 17,036 | |||||
| Loss before the provision for income taxes | (4,024,222 | ) | (1,985,234 | ) | ||||
| PROVISION FOR INCOME TAXES | - | - | ||||||
| NET LOSS | $ | (4,024,222 | ) | $ | (1,985,234 | ) | ||
| NET LOSS PER SHARE | ||||||||
| Basic | $ | (0.03 | ) | $ | (0.02 | ) | ||
| Diluted | $ | (0.03 | ) | $ | (0.02 | ) | ||
| SHARES USED IN CALCULATION OF NET LOSS PER SHARE | ||||||||
| Basic | 127,304,856 | 99,951,385 | ||||||
| Diluted | 127,304,856 | 99,951,385 | ||||||
The accompanying notes are an integral part of these Consolidated Financial Statements.
| F-4 |
CONSOLIDATED STATEMENT OF CHANGES IN STOCKHOLDERS’ DEFICIT
Years ended December 31, 2018 and 2017
| Common Stock | Additional Paid-In- | Deferred | Accumulated | |||||||||||||||||||||
| Shares | Amount | Capital | Compensation | Deficit | Total | |||||||||||||||||||
| Balance at January 1, 2017 | 85,068,709 | $ | 85,069 | $ | 51,963,269 | $ | - | $ | (55,933,862 | ) | $ | (3,885,524 | ) | |||||||||||
| Common stock grants to independent directors | 793,025 | 793 | 149,207 | - | - | 150,000 | ||||||||||||||||||
| Common stock issuance to institutional investor | 567,644 | 568 | 59,432 | - | - | 60,000 | ||||||||||||||||||
| Restricted stock issuances | 34,107,883 | 34,108 | 4,044,327 | - | - | 4,078,435 | ||||||||||||||||||
| Restricted stock issuance to a broker for fees | 558,750 | 559 | 44,141 | - | - | 44,700 | ||||||||||||||||||
| Stock option exercise | 645,288 | 645 | (645 | ) | - | - | - | |||||||||||||||||
| Warrant exercise | 733,217 | 733 | 39,267 | - | - | 40,000 | ||||||||||||||||||
| Deferred compensation | 200,000 | 200 | 40,300 | (10,125 | ) | - | 30,375 | |||||||||||||||||
| Stock based compensation - options | - | - | 61,771 | - | - | 61,771 | ||||||||||||||||||
| Net loss | - | - | - | - | (1,985,234 | ) | (1,985,234 | ) | ||||||||||||||||
| Balance at December 31, 2017 | 122,674,516 | 122,675 | 56,401,069 | (10,125 | ) | (57,919,096 | ) | (1,405,477 | ) | |||||||||||||||
| Common stock grants to independent directors | 1,344,274 | 1,345 | 286,155 | - | - | 287,500 | ||||||||||||||||||
| Warrant Exchange Offering, net | 9,600,286 | 9,600 | 1,234,437 | - | - | 1,244,037 | ||||||||||||||||||
| Common stock grants to service providers | 112,500 | 112 | 22,388 | - | - | 22,500 | ||||||||||||||||||
| Stock option exercise | 156,997 | 157 | (157 | ) | - | - | - | |||||||||||||||||
| Deferred compensation | - | - | - | 10,125 | - | 10,125 | ||||||||||||||||||
| Stock based compensation - options | - | - | 330,146 | - | - | 330,146 | ||||||||||||||||||
| Net loss | - | - | - | - | (4,024,222 | ) | (4,024,222 | ) | ||||||||||||||||
| Balance at December 31, 2018 | 133,888,573 | $ | 133,889 | $ | 58,274,038 | $ | - | $ | (61,943,318 | ) | $ | (3,535,391 | ) | |||||||||||
The accompanying notes are an integral part of these Consolidated Financial Statements.
| F-5 |
CONSOLIDATED STATEMENTS OF CASH FLOWS
For the years ended December 31,
| 2018 | 2017 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net loss | $ | (4,024,222 | ) | $ | (1,985,234 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | 30,570 | 29,422 | ||||||
| Stock based compensation | 650,271 | 242,146 | ||||||
| Bad debt expense on note receivable and accrued interest | 89,933 | - | ||||||
| Changes in assets and liabilities: | ||||||||
| Accounts receivable | 181,960 | (37,243 | ) | |||||
| Inventories | 97,736 | (329,598 | ) | |||||
| Deposits and other assets | (118,168 | ) | 32,045 | |||||
| Prepaid expenses | (1,245 | ) | (2,919 | ) | ||||
| Accrued payroll and payroll related expenses | 39,421 | (20,239 | ) | |||||
| Accounts payable and accrued expenses | (146,784 | ) | (9,003 | ) | ||||
| Net cash used in operating activities | (3,200,528 | ) | (2,080,623 | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||
| Increase in intangible assets | (36,593 | ) | (19,408 | ) | ||||
| Net cash used in investing activities | (36,593 | ) | (19,408 | ) | ||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Proceeds from the issuance of common stock | 1,244,037 | 4,138,435 | ||||||
| Proceeds from the exercise of warrants | - | 40,000 | ||||||
| Net cash provided by financing activities | 1,244,037 | 4,178,435 | ||||||
| NET (DECREASE) INCREASE IN CASH | (1,993,084 | ) | 2,078,404 | |||||
| BEGINNING OF YEAR | 2,236,837 | 158,433 | ||||||
| END OF THE YEAR | $ | 243,753 | $ | 2,236,837 | ||||
| SUPPLEMENTAL DISCLOSURES: | ||||||||
| Cash paid for interest | $ | 4,227 | $ | 3,537 | ||||
| Cash paid for income taxes | $ | - | $ | - | ||||
| NON-CASH INVESTING AND FINANCING ACTIVITIES: | ||||||||
| Conversion of accounts payable into restricted stock | $ | - | $ | 44,700 | ||||
| Settlement of receivables with payables | $ | 301,799 | $ | - | ||||
| Purchases of inventory in accounts payable | $ | 1,237,691 | $ | - | ||||
The accompanying notes are an integral part of these Consolidated Financial Statements.
| F-6 |
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS
NOTE 1 – COMPANY BACKGROUND
The Company’s predecessor, Cardax Pharmaceuticals, Inc. (“Holdings”), was incorporated in the State of Delaware on March 23, 2006.
Cardax, Inc. (the “Company”) (OTCQB:CDXI) is a biopharmaceutical company engaged in the development and commercialization of dietary supplements for inflammatory health and pharmaceuticals for chronic diseases driven by inflammation and oxidative stress. The Company’s first commercial product, ZanthoSyn®, is a physician recommended anti-inflammatory supplement for health and longevity that provides astaxanthin with enhanced absorption and purity. The Company sells ZanthoSyn® primarily through wholesale and e-commerce channels. The Company is also developing CDX-101 (astaxanthin pharmaceutical candidate) and CDX-301 (zeaxanthin pharmaceutical candidate) for pharmaceutical applications. The safety and efficacy of the Company’s products have not been directly evaluated in clinical trials or confirmed by the FDA.
Going concern matters
The accompanying consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. As shown in the accompanying consolidated financial statements, the Company incurred net losses of $4,024,222 and $1,985,234 for the years ended December 31, 2018 and 2017, respectively. The Company has incurred losses since inception resulting in an accumulated deficit of $61,943,318 as of December 31, 2018, and has had negative cash flows from operating activities since inception. The Company expects that its marketing program for ZanthoSyn® will continue to focus on outreach to physicians, healthcare professionals, retail personnel, and consumers, and anticipates further losses in the development of its consumer business. The Company also plans to advance the research and development of its pharmaceutical candidates and anticipates further losses in the development of its pharmaceutical business. As a result of these and other factors, management has determined there is substantial doubt about the Company’s ability to continue as a going concern.
During the year ended December 31, 2018, the Company raised additional capital to carry out its business plan. As part of the Company’s efforts, it raised an additional $1.44 million in gross proceeds through the exchange of 9.6 million warrants via a warrant exchange offering that closed on July 27, 2018. Stock issuance costs associated with this capital raise totaled $196,006, for a net total $1,244,037 raised in this offering.
The Company’s continued ability to raise additional capital through future equity and debt securities issuances is unknown. Obtaining additional financing, the successful development of the Company’s contemplated plan of operations, and its transition, ultimately, to profitable operations are necessary for the Company to continue operations. The ability to successfully resolve these factors raises substantial doubt about the Company’s ability to continue as a going concern. The consolidated financial statements of the Company do not include any adjustments that may result from the outcome of these uncertainties.
| F-7 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of presentation
The consolidated financial statements have been consistently prepared in accordance with accounting principles generally accepted in the United States (“U.S. GAAP”) and include the accounts of Cardax, Inc., and its wholly owned subsidiary, Cardax Pharma, Inc., and its predecessor, Cardax Pharmaceuticals, Inc., which was merged with and into Cardax, Inc. All significant intercompany balances and transactions have been eliminated in consolidation.
Use of estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in our consolidated financial statements and the accompanying notes. Estimates in these consolidated financial statements include asset valuations, estimates of future cash flows from and the economic useful lives of long-lived assets, valuations of stock compensation, certain accrued liabilities, income taxes and tax valuation allowances, and fair value estimates. Despite management’s intention to establish accurate estimates and reasonable assumptions, actual results could differ materially from these estimates and assumptions.
Cash
The Company considers all highly liquid investments with maturities of three months or less at the time of purchase to be cash equivalents. The Company held no cash equivalents as of December 31, 2018 and 2017.
The Company maintains cash deposit accounts at one financial institution. Accounts at this institution are insured by the Federal Deposit Insurance Corporation up to $250,000. The Company’s cash balance at times may exceed these limits. As of December 31, 2018 and 2017, the Company had $0 and $1,988,139, respectively, in excess of federally insured limits on deposit.
Accounts receivable
Accounts receivable of $157,082 and $37,243 as of December 31, 2018 and 2017, respectively, consists of amounts due from sales of dietary supplements.
It is the Company’s policy to provide for an allowance for doubtful collections based upon a review of outstanding receivables, historical collection information, and existing economic conditions. Normal receivables are due 60 days after the issuance of the invoice. Receivables past due more than 90 days are considered delinquent. Delinquent receivables are written off based on individual credit evaluation and specific circumstances of the customer. There was no allowance necessary as of December 31, 2018 and 2017.
| F-8 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
Inventories
Inventories are stated at the lower of cost or net realizable value. Cost is determined using the average cost method. Net realizable value is the estimated selling prices in the ordinary course of business, less reasonably predictable costs of completion, disposal, and transportation. Inventory costs include third party costs for finished goods. The Company utilizes contract manufacturers and receives inventory in finished form.
The Company provides a reserve against inventory for known or expected inventory obsolescence. The reserve is determined by specific review of inventory items for product age and quality that may affect salability. There were no reserves necessary for inventory as of December 31, 2018 and 2017.
Property and equipment, net
Property and equipment are recorded at cost, less depreciation. Equipment under capital lease obligations and leasehold improvements are amortized on the straight-line method over the shorter period of the lease term or the estimated useful life of the equipment. Such amortization is included in depreciation and amortization in the consolidated financial statements. Depreciation is calculated using the straight-line method over the estimated useful lives of the respective assets are as follows.
| Furniture and office equipment | 7 years | |
| Research and development equipment | 3 to 7 years | |
| Information technology equipment | 5 years | |
| Software | 3 years |
Major additions and improvements are capitalized, and routine expenditures for repairs and maintenance are charged to expense as incurred. When assets are retired or otherwise disposed of, the cost and related accumulated depreciation are removed from the accounts, and any resulting gain or loss is charged to income for the period.
Impairment of long-lived assets
The Company evaluates long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset or group of assets, as appropriate, may not be recoverable.
When the sum of the undiscounted future net cash flows expected to result from the use and the eventual disposition is less than the carrying amounts, an impairment loss would be measured based on the discounted cash flows compared to the carrying amounts. There was no impairment charge recorded for the years ended December 31, 2018 and 2017.
| F-9 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
Revenue from contracts with customers
In May 2014, the Financial Accounting Standards Board (“FASB”) issued a new standard related to revenue recognition. Under the standard, revenue is recognized when a customer obtains control of promised goods or services in an amount that reflects the consideration the entity expects to receive in exchange for those goods or services. In addition, the standard requires disclosure of the nature, amount, timing, and uncertainty of revenue and cash flows arising from contracts with customers.
The Company adopted this standard effective January 1, 2018, using the retrospective method. As there was no impact on contracts that were previously completed and no significant impact to contracts completed after adoption, there was no need to restate prior results from operations.
The Company recognizes revenues from its contracts with customers for its products through wholesale and e-commerce channels when goods and services have been identified, the payment terms agreed to, the contract has commercial substance, both parties have approved the contract, and it is probable that the Company will collect all substantial consideration.
The following table presents our revenues disaggregated by revenue source and geographical location. Sales and usage-based taxes are included as a component of revenues for the years ended December 31:
| Geographical area | Source | 2018 | 2017 | |||||||||
| United States | Nutraceuticals | $ | 1,494,462 | $ | 610,323 | |||||||
| Hong Kong | Nutraceuticals | $ | 16,413 | $ | - | |||||||
Sales discounts, rebates, promotional amounts to vendors, and returns and allowances are recorded as a reduction to sales in the period in which sales are recorded. Sales discounts and other adjustments are recorded at the time of sale.
Cost of goods sold
Cost of goods sold is comprised of costs to manufacture or acquire products sold to customers, direct and indirect distribution costs, and other costs incurred in the sale of goods.
Shipping and handling costs
Shipping and handling costs are included in cost of goods sold. Shipping and handling costs were $21,603 and $10,366 for the years ended December 31, 2018 and 2017, respectively.
Sales and use tax
Revenues, as presented on the accompanying income statement, include taxes collected from customers and remitted to governmental authorities. Such taxes were $3,329 and $5,132 for the years ended December 31, 2018 and 2017, respectively.
| F-10 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
Research and development
Research and development costs are expensed as incurred and consists primarily of manufacturing of products, third-party research, laboratory supplies, and scientific advisory boards. The focus of these costs is on the development of astaxanthin, zeaxanthin, and related compounds. For the years ended December 31, 2018 and 2017, research and development costs were $269,077 and $97,479, respectively.
Advertising
Advertising costs are expensed as incurred and are included as an element of sales and marketing costs in the accompanying consolidated statements of operations. For the years ended December 31, 2018 and 2017, advertising costs were $364,306 and $84,317, respectively.
Income taxes
The Company accounts for income taxes under an asset and liability approach. Deferred income taxes reflect the impact of temporary differences between assets and liabilities recognized for financial reporting purposes and the amounts recognized for income tax reporting purposes, net operating loss carry-forwards, and other tax credits measured by applying currently enacted tax laws. A valuation allowance is provided when necessary to reduce deferred tax assets to an amount that is more likely than not to be realized.
The Company determines whether a tax position is more likely than not to be sustained upon examination, including resolution of any related appeals or litigation processes, based on the technical merits of the position. The Company uses a two-step approach to recognizing and measuring uncertain tax positions. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates that it is more likely than not that the position will be sustained upon tax authority examination, including resolution of related appeals or litigation processes, if any. The second step is to measure the tax benefit as the largest amount that is more than 50% likely of being realized upon ultimate settlement.
The Company files income tax returns in the United States (“U.S.”) Federal and the States of Hawaii and California jurisdictions. Tax regulations within each jurisdiction are subject to the interpretation of the related tax laws and regulations and require significant judgment to apply. The federal and state income tax returns of the Company are subject to examination by the IRS and state taxing authorities, generally for three years after they were filed.
The Company did not recognize any tax liabilities for income taxes associated with unrecognized tax benefits as of December 31, 2018 and 2017. The Company’s policy is to include interest and penalties related to unrecognized tax benefits, if any, within the provision for income taxes in the consolidated statements of operations.
In 2017, the Company adopted FASB issued Accounting Standards Update (“ASU”) No. 2015-17, Income Taxes (Topic 740). This ASU was issued as part of the FASB’s simplification initiative focused on improving areas of U.S. GAAP for which cost and complexity may be reduced while maintaining or improving the usefulness of information disclosed within the financial statements. ASU No. 2015-17 simplifies the presentation of deferred income taxes by requiring that deferred tax liabilities and assets be presented net and classified as noncurrent in a classified statement of financial position. As a result of this adoption, the Company now presents deferred tax assets as a single line item, net, in long-term assets or labilities.
| F-11 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
Fair value measurements
U.S. GAAP establishes a framework for measuring fair value. That framework provides a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements) and the lowest priority to unobservable inputs (Level 3 measurements).
The three levels of the fair value hierarchy are described below:
| Level 1: | Inputs to the valuation methodology are unadjusted quoted prices for identical assets or liabilities in active markets that the Company has the ability to access. | |
| Level 2: | Inputs to the valuation methodology include: |
| ● | Quoted prices for similar assets or liabilities in active markets; | |
| ● | Quoted prices for identical or similar assets or liabilities in inactive markets; | |
| ● | Inputs other than quoted prices that are observable for the asset or liability; and | |
| ● | Inputs that are derived principally from or corroborated by observable market data by correlation or other means. |
If the asset or liability has a specified (contractual) term, the Level 2 input must be observable for substantially the full term of the asset or liability.
| Level 3: | Inputs to the valuation methodology are unobservable and significant to the fair value measurement. |
The asset’s or liability’s fair value measurement level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value measurement. Valuation techniques used need to maximize the use of observable inputs and minimize the use of unobservable inputs.
As of December 31, 2018 and 2017, there were no recurring fair value measurements of assets and liabilities subsequent to initial recognition.
| F-12 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
Stock based compensation
The Company accounts for stock-based compensation costs under the provisions of FASB’s Accounting Standards Codification (“ASC”) No. 718, Compensation—Stock Compensation and ASC No. 505, Equity, which requires the measurement and recognition of compensation expense related to the fair value of stock based compensation awards that are ultimately expected to vest. Stock based compensation expense recognized includes the compensation cost for all stock-based payments granted to employees, officers, directors, and consultants based on the grant date fair value estimated. These standards also apply to awards modified, repurchased, or canceled during the periods reported.
Basic and diluted net loss per share
Basic earnings per common share is calculated by dividing net loss for the year by the weighted average number of common shares outstanding during the year. Diluted earnings per common share is calculated by dividing net loss for the year by the sum of the weighted average number of common shares outstanding during the year plus the number of potentially dilutive common shares (“dilutive securities”) that were outstanding during the year. Dilutive securities include options granted pursuant to the Company’s stock option plans, and warrants issued to non-employees. Potentially dilutive securities are excluded from the computation of earnings per share in periods in which a net loss is reported, as their effect would be antidilutive.
Recently issued accounting pronouncements
In February 2016, the FASB issued ASU No. 2016-02, Leases. This ASU requires management to recognize lease assets and lease liabilities for all leases. ASU No. 2016-02 retains a distinction between finance leases and operating leases. The classification criteria for distinguishing between finance leases and operating leases are substantially similar to the classification criteria for distinguishing between capital leases and operating leases in the previous lease guidance. The result of retaining a distinction between finance leases and operating leases is that under the lessee accounting model, the effect of leases in the statement of comprehensive income and the statement of cash flows is largely unchanged from previous U.S. GAAP. The guidance in ASU No. 2016-02 is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. The Company applied the modified retrospective approach in adopting this standard. The modified retrospective approach includes a number of optional practical expedients that the Company elected to apply; primarily the identification and classification of leases that commenced before the effective date, initial direct costs for leases that commenced before the effective date, and the ability to use hindsight in evaluating lessee options to extend or terminate a lease or to purchase the underlying asset. As part of this adoption, the Company will, in effect, continue to account for leases that commence before the effective date in accordance with previous GAAP unless the lease is modified, except that lessees are required to recognize a right-of-use asset and a lease liability for all operating leases at each reporting date based on the present value of the remaining minimum rental payments that were tracked and disclosed under previous GAAP. This adoption of this standard on January 1, 2019, resulted in the Company recognizing a right-to-use asset and lease liability of approximately $35,000. The Company elected to not recognize any right-to-use assets or liabilities for leases that are twelve months or less. Lease costs are recognized straight-line over the term of the lease. The adoption of this standard did not impact retained earnings of cash flows of the Company.
In June 2018, the FASB issued ASU No. 2018-07, Compensation – Stock Compensation (Topic 718), Improvements to Nonemployee Share-Based Payment Accounting. This ASU is intended to simplify aspects of share-based compensation issued to non-employees by making the guidance consistent accounting for employee share-based compensation. The guidance in ASU No. 2018-07 is effective for annual reporting periods, and interim periods within those years, beginning after December 15, 2018. The Company is currently in the process of evaluating the impact of the adoption of this ASU on its consolidated financial statements.
| F-13 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
Recently issued accounting pronouncements
In August 2018, the FASB issued ASU No. 2018-13, Fair Value Measurement. This ASU modifies the disclosure requirements on fair value measurements in Topic 820, Fair Value Measurement, based on the concepts in the FASB’s Concepts Statement, including the consideration of costs and benefits. The guidance in ASU No. 2018-13 is effective for annual reporting periods, and interim periods within those years, beginning after December 15, 2019. The Company is currently in the process of evaluating the impact of the adoption of this ASU on its consolidated financial statements.
The Company does not believe that any other recently issued, but not yet effective accounting pronouncements, if adopted, would have a material effect on the consolidated financial statements.
Reclassifications
The Company has made certain reclassifications to conform its prior periods’ data to the current presentation. These reclassifications had no effect on the reported results of operations or cash flows.
| F-14 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 3 – INVENTORIES
Inventories consist of the following as of December 31:
| 2018 | 2017 | |||||||
| Finished goods | $ | 96,750 | $ | 240,917 | ||||
| Raw materials | 1,383,630 | 98,937 | ||||||
| Packing supplies and materials | - | 571 | ||||||
| Total inventories | $ | 1,480,380 | $ | 340,425 | ||||
As of December 31, 2018, $1,383,630 in raw materials were held at the manufacturer’s facility for future production.
NOTE 4 – PROPERTY AND EQUIPMENT, net
Property and equipment, net, consists of the following as of December 31:
| 2018 | 2017 | |||||||
| Information technology equipment | $ | - | $ | 31,892 | ||||
| Less accumulated depreciation | - | (29,991 | ) | |||||
| Total property and equipment, net | $ | - | $ | 1,901 | ||||
Depreciation expense was $1,901 and $5,854 for the years ended December 31, 2018 and 2017, respectively.
During the year ended December 31, 2018, the Company wrote off its fully depreciated equipment. There was no gain or loss recognized for this write-off.
NOTE 5 – INTANGIBLE ASSETS, net
Intangible assets, net, consists of the following as of December 31:
| 2018 | 2017 | |||||||
| Patents | $ | 578,326 | $ | 493,027 | ||||
| Less accumulated amortization | (292,512 | ) | (263,843 | ) | ||||
| 285,814 | 229,184 | |||||||
| Patents pending | 148,720 | 197,426 | ||||||
| Total intangible assets, net | $ | 434,534 | $ | 426,610 | ||||
Patents are amortized straight-line over a period of fifteen years. Amortization expense was $28,669 and $23,568 for the years ended December 31, 2018 and 2017, respectively.
The Company has capitalized costs for several patents that are still pending. In those instances, the Company has not recorded any amortization. The Company will commence amortization when these patents are approved.
The Company owns 28 issued patents, including 14 in the United States and 14 others in Europe, Canada, China, India, Japan, and Hong Kong. These patents will expire beginning in 2023 through 2028, subject to any patent term extensions of the individual patent. The Company has 1 patent application pending in the United States and 2 patent applications pending in Europe and Brazil.
| F-15 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 6 –ACCRUED SEPARATION COSTS
On August 9, 2016, the Company entered into a separation agreement with an employee to pay $118,635 of accrued compensation over nine-years. This amount is included in accrued payroll and payroll related expenses in the accompanying consolidated balance sheets. This amount does not yield interest and matures as follows for the years ended December 31:
| 2019 | $ | 9,000 | ||
| 2020 | 9,000 | |||
| 2021 | 12,000 | |||
| 2022 | 12,000 | |||
| 2023 | 18,000 | |||
| Thereafter | 41,635 | |||
| 101,635 | ||||
| Less current portion | (9,000 | ) | ||
| $ | 92,635 |
NOTE 7 – STOCKHOLDERS’ DEFICIT
Warrant exchange offering
In June 2018, the Company commenced an offering to exchange outstanding warrants for shares of common stock under a Form S-4 Registration Statement. These shares of common stock were issued to warrant holders in exchange for (i) their outstanding warrants to purchase shares of common stock at $0.625 per share, and (ii) cash payment of $0.15 per share. This offering closed on July 27, 2018, and resulted in an exchange of 9.6 million warrants and $1,440,043 in gross proceeds for 9,600,286 shares of common stock. Stock issuance costs associated with this capital raise totaled $196,006, resulting in a net total of $1,244,037 raised in this offering.
Self-directed stock issuance
During the year ended December 31, 2017, the Company sold securities in a self-directed offering in the aggregate amount of $179,000, $3,774,456, and $124,979 at $0.08, $0.12, and $0.30, respectively, per unit. Each $0.08 unit consisted of 1 share of restricted common stock (2,237,500 shares), a five-year warrant to purchase 1 share of restricted common stock (2,237,500 warrant shares) at $0.08 per share, a five-year warrant to purchase 1 share of restricted common stock (2,237,500 warrant shares) at $0.12 per share, and a five-year warrant to purchase 1 share of restricted common stock (2,237,500 warrant shares) at $0.16 per share. Each $0.12 unit consisted of 1 share of restricted common stock (31,453,788 shares) and a five-year warrant to purchase 1 share of restricted common stock (31,453,788 warrant shares) at $0.12 per share. Each $0.30 unit consisted of 1 share of restricted common stock (416,595 shares) and a five-year warrant to purchase 1 share of restricted common stock (416,595 warrant shares) at $0.30 per share.
| F-16 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 7 – STOCKHOLDERS’ DEFICIT (continued)
Equity purchase agreement
During the year ended December 31, 2017, the Company sold 567,644 shares of common stock for $60,000, pursuant to an equity purchase agreement.
Payable settlement
In May 2017, the Company settled a payable in the amount of $44,700 with a previously engaged broker dealer through the issuance of securities at $0.08 per unit. Each unit consisted of 1 share of restricted common stock (558,750 shares), a five-year warrant to purchase 1 share of restricted common stock (558,750 warrant shares) at $0.08 per share, a five-year warrant to purchase 1 share of restricted common stock (558,750 warrant shares) at $0.12 per share, and a five-year warrant to purchase 1 share of restricted common stock (558,750 warrant shares) at $0.16 per share.
Shares outstanding
As of December 31, 2018 and 2017, the Company had a total of 133,888,573 and 122,674,516 shares of common stock outstanding.
NOTE 8 – STOCK GRANTS
Director stock grants
During 2018 and 2017, the Company granted its independent directors an aggregate of 1,344,274 and 793,025, respectively, shares of restricted common stock in the Company. These shares were fully vested upon issuance. The increase in number of shares issued was due to the expansion of the Board of Directors by two members in June 2018. The expense recognized for these grants based on the grant date fair value was $287,500 and $150,000 for the years ended December 31, 2018 and 2017, respectively.
Consultant stock grants
On April 10, 2017, the Company granted a consultant 100,000 shares of restricted common stock valued at $0.23 per share. These shares are subject to a risk of forfeiture and vest quarterly in arrears commencing on April 1, 2017. The Company recognized $17,250 in stock-based compensation related to this grant during the year ended December 31, 2017.
On August 8, 2017, the Company granted a consultant 100,000 shares of restricted common stock valued at $0.175 per share. These shares are subject to a risk of forfeiture and vest 25% upon grant and quarterly in arrears thereafter commencing on September 1, 2017. The Company recognized $10,125 and $13,125 in stock-based compensation related to this grant during the years ended December 31, 2018 and 2017.
On December 31, 2018, the Company granted consultants 112,500 shares of restricted common stock valued at $0.20 per share. These shares were fully vested upon issuance. The Company recognized $22,500 in stock-based compensation related to these grants during the year ended December 31, 2018.
| F-17 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 9 – STOCK OPTION PLANS
On February 7, 2014, the Company adopted the 2014 Equity Compensation Plan. Under this plan, the Company may issue options to purchase shares of common stock to employees, directors, advisors, and consultants. The aggregate number of shares that may be issued under this plan upon adoption was 30,420,148. On April 16, 2015, the majority stockholder of the Company approved an increase in the Company’s 2014 Equity Compensation Plan by 15 million shares, which was subsequently increased in December 2018 by an additional 5 million shares to allow for a total of 50,420,148 shares available under the plan.
Under the terms of the 2014 Equity Compensation Plan and the 2006 Stock Incentive Plan (collectively, the “Plans”), incentive stock options may be granted to employees at a price per share not less than 100% of the fair market value at date of grant. If the incentive stock option is granted to a 10% stockholder, then the purchase or exercise price per share shall not be less than 110% of the fair market value per share of common stock on the grant date. Non-statutory stock options and restricted stock may be granted to employees, directors, advisors, and consultants at a price per share, not less than 100% of the fair market value at date of grant. Options granted are exercisable, unless specified differently in the grant documents, over a default term of ten years from the date of grant and generally vest over a period of four years.
A summary of stock option activity is as follows:
| Options | Weighted average exercise price | Weighted | Aggregate intrinsic value | |||||||||||||
| Outstanding January 1, 2017 | 36,821,969 | $ | 0.41 | 5.94 | $ | 301,273 | ||||||||||
| Exercisable January 1, 2017 | 36,771,969 | $ | 0.41 | 5.94 | $ | 299,273 | ||||||||||
| Canceled | - | |||||||||||||||
| Granted | 2,161,458 | |||||||||||||||
| Exercised | (770,000 | ) | ||||||||||||||
| Forfeited | - | |||||||||||||||
| Outstanding December 31, 2017 | 38,213,427 | $ | 0.41 | 5.23 | $ | 562,456 | ||||||||||
| Exercisable December 31, 2017 | 36,213,427 | $ | 0.41 | 4.98 | $ | 562,456 | ||||||||||
| Canceled | (350,000 | ) | ||||||||||||||
| Granted | 2,833,334 | |||||||||||||||
| Exercised | (200,000 | ) | ||||||||||||||
| Forfeited | - | |||||||||||||||
| Outstanding December 31, 2018 | 40,496,761 | $ | 0.40 | 4.52 | $ | 986,808 | ||||||||||
| Exercisable December 31, 2018 | 37,157,179 | $ | 0.41 | 4.10 | $ | 966,808 | ||||||||||
| F-18 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 9 – STOCK OPTION PLANS (continued)
The aggregate intrinsic value in the table above is before applicable income taxes and represents the excess amount over the exercise price option recipients would have received if all options had been exercised on December 31, 2018, based on a valuation of the Company’s stock for that day.
A summary of the Company’s non-vested options for the years ended December 31, 2018 and 2017 are presented below:
| Non-vested at January 1, 2017 | 50,000 | |||
| Granted | 2,161,458 | |||
| Vested | (211,458 | ) | ||
| Canceled | - | |||
| Non-vested at December 31, 2017 | 2,000,000 | |||
| Granted | 2,833,334 | |||
| Vested | (1,143,752 | ) | ||
| Canceled | (350,000 | ) | ||
| Non-vested at December 31, 2018 | 3,339,582 |
The Company estimates the fair value of stock options granted on each grant date using the Black-Scholes option valuation model and recognizes an expense ratably over the requisite service period. The range of fair value assumptions related to options issued were as follows for the years ended December 31:
| 2018 | 2017 | |||||||
| Dividend yield | 0.0 | % | 0.0 | % | ||||
| Risk-free rate | 2.38% - 3.04 | % | 1.89% - 2.26 | % | ||||
| Expected volatility | 214% - 226 | % | 221% - 232 | % | ||||
| Expected term | 3 - 7 years | 5 - 7 years | ||||||
The expected volatility was calculated based on the historical volatility of the Company. The risk-free interest rate used was based on the U.S. Treasury constant maturity rate in effect at the time of grant for the expected term of the stock options to be valued. The expected dividend yield was zero, because the Company does not anticipate paying a dividend within the relevant timeframe. Due to a lack of historical information needed to estimate the Company’s expected term, it was estimated using the simplified method allowed.
The Company records forfeitures as they occur and reverses compensation cost previously recognized, in the period the award is forfeited, for an award that is forfeited before completion of the requisite service period.
| F-19 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 9 – STOCK OPTION PLANS (continued)
Stock option exercise
During the year ended December 31, 2018, the Company issued 156,997 shares of common stock in connection with the cashless exercise of stock options for 100,000, 50,000, and 50,000 shares of common stock exercisable at $0.06 per share with 43,003 shares of common stock withheld with an aggregate fair market value equal to the aggregate exercise price.
During the year ended December 31, 2017, the Company issued 645,288 shares of common stock in connection with the cashless exercise of stock options for 100,000, 45,000, and 625,000 shares of common stock exercisable at $0.155, $0.06, and $0.06, respectively, per share with 124,712 shares of common stock withheld with an aggregate fair market value equal to the aggregate exercise price.
The Company recognized stock-based compensation expense related to options during the:
| Years ended December 31 | ||||||||
| 2018 | 2017 | |||||||
| Amount | Amount | |||||||
| Service provider compensation | $ | 124,896 | $ | 3,500 | ||||
| Employee compensation | 205,250 | 33,271 | ||||||
| Director compensation | - | 25,000 | ||||||
| Total | $ | 330,146 | $ | 61,771 | ||||
| F-20 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 10 – WARRANTS
The following is a summary of the Company’s warrant activity:
| Warrants | Weighted average exercise price | Weighted average remaining contractual term in years | Aggregate intrinsic value | |||||||||||||
| Outstanding January 1, 2017 | 88,365,036 | $ | 0.30 | 3.50 | $ | 543,770 | ||||||||||
| Exercisable January 1, 2017 | 88,365,036 | $ | 0.30 | 3.50 | $ | 543,770 | ||||||||||
| Canceled | - | |||||||||||||||
| Granted | 40,259,133 | |||||||||||||||
| Exercised | (798,000 | ) | ||||||||||||||
| Forfeited | (392,047 | ) | ||||||||||||||
| Outstanding December 31, 2017 | 127,434,122 | $ | 0.24 | 3.15 | $ | 3,957,689 | ||||||||||
| Exercisable December 31, 2017 | 127,434,122 | $ | 0.24 | 3.15 | $ | 3,957,689 | ||||||||||
| Canceled | - | |||||||||||||||
| Granted | 315,010 | |||||||||||||||
| Exercised | (9,600,286 | ) | ||||||||||||||
| Forfeited | (101,984 | ) | ||||||||||||||
| Outstanding December 31, 2018 | 118,046,862 | $ | 0.20 | 2.32 | $ | 7,848,637 | ||||||||||
| Exercisable December 31, 2018 | 118,046,862 | $ | 0.20 | 2.32 | $ | 7,848,637 | ||||||||||
The Company estimates the fair value of warrants granted on each grant date using the Black-Scholes option valuation model. The expected volatility is calculated based on the historical volatility of the Company. The risk-free interest rate used is based on the U.S. Treasury constant maturity rate in effect at the time of grant for the expected term of the warrants to be valued. The expected dividend yield is zero, because the Company does not anticipate paying a dividend within the relevant timeframe. Due to a lack of historical information needed to estimate the Company’s expected term, it is estimated using the simplified method allowed.
The Company did not recognize any stock-based compensation expense related to warrants during the years ended December 31, 2018 and 2017, respectively.
Warrant exercise
During the year ended December 31, 2017, the Company issued 233,217 shares of common stock in connection with the cashless exercise of a warrant for 298,000 shares of common stock at $0.10 per share with 64,783 shares of common stock withheld with an aggregate fair market value equal to the aggregate exercise price.
During the year ended December 31, 2017, the Company issued 500,000 shares of common stock in connection with the exercise of a warrant for 500,000 shares of common stock at $0.08 per share in exchange for $40,000.
| F-21 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 10 – WARRANTS (continued)
Warrant exchange offering
In June 2018, the Company commenced an offering to exchange outstanding warrants for shares of common stock under a Form S-4 Registration Statement. These shares of common stock were issued to warrant holders in exchange for (i) their outstanding warrants to purchase shares of common stock at $0.625 per share, and (ii) cash payment of $0.15 per share. This offering closed on July 27, 2018, and resulted in an exchange of 9.6 million warrants and $1,440,043 in gross proceeds for 9,600,286 shares of common stock. Stock issuance costs associated with this capital raise totaled $196,006, resulting in a net total of $1,244,037 raised in this offering. As part of this offering, warrants to purchase 315,010 shares of common stock at $0.21 per share were issued to investment bankers for their services.
Warrant expiration
During the years ended December 31, 2018 and 2017, warrants to purchase an aggregate of 101,984 and 392,047 shares of restricted common stock expired.
NOTE 11 – INCOME TAXES
The following table presents a reconciliation of the statutory Federal rate and the Company’s effective tax rate for the years ended December 31:
| 2018 | 2017 | |||||||
| Tax provision (benefit) at Federal statutory rate | (21.00 | )% | (34.00 | )% | ||||
| Accrued compensation | (0.28 | )% | (0.32 | )% | ||||
| Stock based compensation | 3.37 | % | 4.15 | % | ||||
| Depreciation and amortization | 0.15 | % | 0.59 | % | ||||
| Other | 0.07 | % | 0.26 | % | ||||
| Change in valuation allowance | 17.69 | % | 29.32 | % | ||||
| Effective tax rate | 0.00 | % | 0.00 | % | ||||
The effective tax rate for the three and years ended December 31, 2018 and 2017, differs from the statutory rate of 21% and 34% for the years ended December 31, 2018 and 2017, respectively, as a result of state taxes (net of Federal benefit), permanent differences, and a reserve against deferred tax assets.
There was not a provision for income taxes for the years ended December 31, 2018 and 2017.
| F-22 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 11 – INCOME TAXES (continued)
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The following table presents significant components of the Company’s deferred tax assets and liabilities for the years ended December 31:
| 2018 | 2017 | |||||||
| DEFERRED TAX ASSETS, net: | ||||||||
| Net operating loss carryforwards | $ | 9,633,893 | $ | 8,705,467 | ||||
| Accrued compensation | 1,080,432 | 1,074,903 | ||||||
| Stock based compensation | 178,174 | 66,348 | ||||||
| Credit carryforwards | 52,592 | 71,910 | ||||||
| Depreciation and amortization carryforwards | (63,917 | ) | (71,054 | ) | ||||
| Total | 10,881,174 | 9,847,574 | ||||||
| Less valuation allowance | (10,881,174 | ) | (9,847,574 | ) | ||||
| NET DEFERRED TAX ASSETS | $ | - | $ | - | ||||
As of December 31, 2018, the Company had a Federal net operating loss carryforward of $36,950,157. In addition, the Company had a net operating loss carryforward for Hawaii income tax purposes of $29,286,880 as of December 31, 2018. These amounts may be used to offset up to 80% of future taxable income and differ from the Company’s accumulated deficit due to permanent and temporary tax differences.
The Company’s valuation allowance was primarily related to the operating losses. The valuation allowance is determined in accordance with the provisions of ASC No. 740, Income Taxes, which requires an assessment of both negative and positive evidence when measuring the need for a valuation allowance. Based on the available objective evidence and the Company’s history of losses, management provides no assurance that the net deferred tax assets will be realized. As of December 31, 2018 and 2017, the Company has applied a valuation allowance against its deferred tax assets net of the expected income from the reversal of the deferred tax liabilities.
Recent tax legislation
On December 22, 2017, the Tax Cuts and Jobs Act (“TCJA”) was enacted into law, which significantly changes existing U.S. tax law and includes numerous provisions that affect our business, such as reducing the U.S. federal statutory tax rate. The TCJA reduces the U.S. federal statutory tax rate from 35% to 21% effective January 1, 2018.
As a result of TCJA, the Company recorded a change in its deferred tax asset of approximately, $3.8 million for the year ended December 31, 2017, which was offset by an adjustment to the allowance.
State tax credits
The Company received a refundable tax credit of $17,253 from the State of Hawaii during the year ended December 31, 2017. This amount is recorded as other income in the consolidated statement of operations.
| F-23 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 12 – BASIC AND DILUTED NET LOSS PER SHARE
The following table sets forth the computation of the Company’s basic and diluted net loss per share for the years ended December 31:
| 2018 | ||||||||||||
| Net
Loss (Numerator) | Shares
(Denominator) | Per
share amount | ||||||||||
| Basic loss per share | $ | (4,024,222 | ) | 127,304,856 | $ | (0.03 | ) | |||||
| Effect of dilutive securities—Common stock options and warrants | - | - | - | |||||||||
| Diluted loss per share | $ | (4,024,222 | ) | 127,304,856 | $ | (0.03 | ) | |||||
| 2017 | ||||||||||||
| Net
Loss (Numerator) | Shares
(Denominator) | Per
share amount | ||||||||||
| Basic loss per share | $ | (1,985,234 | ) | 99,951,385 | $ | (0.02 | ) | |||||
| Effect of dilutive securities—Common stock options and warrants | - | - | - | |||||||||
| Diluted loss per share | $ | (1,985,234 | ) | 99,951,385 | $ | (0.02 | ) | |||||
The following outstanding shares of common stock equivalents were excluded from the computation of diluted net loss per share for the periods presented because including them would have been antidilutive for the years ended December 31:
| 2018 | 2017 | |||||||
| Common stock options | 40,496,761 | 38,213,427 | ||||||
| Common stock warrants | 118,046,862 | 127,434,122 | ||||||
| Total common stock equivalents | 158,543,623 | 165,647,549 | ||||||
| F-24 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 13 – LEASES
Manoa Innovation Center
The Company entered into an automatically renewable month-to-month lease for office space on August 13, 2010. Under the terms of this lease, the Company must provide a written notice 45 days prior to vacating the premises. Total rent expense under this agreement as amended was $39,302 and $29,690, for the years ended December 31, 2018 and 2017, respectively.
Fleet Lease
In January 2018, the Company entered into a vehicle lease arrangement with a rental company for three vehicles. The terms of the leases require monthly payments of $1,619 for three years. These leases convert to month-to-month leases in January 2021 unless terminated. Total lease expense under this agreement was $21,196 for the year ended December 31, 2018.
Future minimum lease payments are as follows for the years ended December 31:
| 2019 | $ | 17,868 | ||
| 2020 | 17,868 | |||
| 2021 | 1,489 | |||
| $ | 37,225 |
NOTE 14 – COMMITMENTS
Patent payable
As part of the formation of the Company, a patent license was transferred to the Company. The original license began in 2006. Under the terms of the license the Company agreed to pay $10,000 per year through 2015 and royalties of 2% on any revenues resulting from the license. There were no revenues generated by this license during the years ended December 31, 2018 and 2017. The remaining obligation of $20,000 as of December 31, 2018 and 2017, is recorded as a part of accounts payable on the consolidated balance sheets. The license expired in February 2016.
Employee settlement
As of December 31, 2018 and 2017, the Company owed a former employee a severance settlement payable in the amount of $50,000 for accrued vacation benefits. As part of the severance settlement, a stock option previously granted to the former employee was fully vested and extended.
| F-25 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 14 – COMMITMENTS (continued)
BASF agreement and license
In November 2006, the Company entered into a joint development and supply agreement with BASF SE (“BASF”). Under the agreement, the Company granted BASF an exclusive world-wide license to the Company’s rights related to the development and commercialization of astaxanthin nutraceutical products; the Company retains all rights related to astaxanthin pharmaceutical products. The Company is to receive specified royalties based on future net sales of such astaxanthin nutraceutical products. No royalties were realized from this agreement during the years ended December 31, 2018 and 2017.
Capsugel agreement
On August 18, 2014, the Company entered into a collaboration agreement with Capsugel US, LLC (“Capsugel”) for the joint commercial development of astaxanthin products (“Capsugel Astaxanthin Products”) for the consumer health market that contain nature-identical synthetic astaxanthin and use Capsugel’s proprietary formulation technology. The agreement provides for the parties to jointly administer activities under a product development plan that will include identifying at least one mutually acceptable third-party marketer who will further develop, market and distribute Capsugel Astaxanthin Products. Capsugel will share revenues with the Company based on net sales of products that are developed under the collaboration. No revenues were realized from this agreement during the years ended December 31, 2018 and 2017.
NOTE 15 – SUBSEQUENT EVENTS
The Company evaluated all material events through the date the financials were ready for issuance and noted the following non-recognized events for disclosure.
On January 11, 2019, the Company entered into a $1,000,000 revolving inventory financing facility with a lender. Use of proceeds from this facility is limited to the purchase of inventory, including raw materials, intermediates, and finished goods, unless otherwise waived by the lender. This facility accrues interest at the rate of 12% per annum, is unsecured, and matures in three years from origination. This facility also requires monthly interest payments. As of March 27, 2019, the aggregate unpaid principal amount under this facility was $1,000,000.
On February 7, 2019, the Company registered 167,730,236 shares of its common stock, par value $0.001 per share, held by the registering stockholders, consisting of (i) 69,115,849 shares of its issued and outstanding common stock, and (ii) 98,614,387 shares of its common stock that may be issued upon the exercise of outstanding warrants.
On February 22, 2019, the Company sold securities in a self-directed offering in the aggregate amount of $20,000 at $0.30 per unit. Each unit consisted of 2 shares of restricted common stock (133,332 shares) and a five-year warrant to purchase 1 share of restricted common stock (66,666 warrant shares) at $0.20 per share.
***
| F-26 |
CONDENSED CONSOLIDATED BALANCE SHEETS
As of
| September 30, 2019 | December 31, 2018 | |||||||
| (Unaudited) | ||||||||
| ASSETS | ||||||||
| CURRENT ASSETS | ||||||||
| Cash | $ | 7,470 | $ | 243,753 | ||||
| Accounts receivable | 185,419 | 157,082 | ||||||
| Inventories | 1,307,727 | 1,480,380 | ||||||
| Deposits and other assets | 119,066 | 119,066 | ||||||
| Prepaid expenses | 45,096 | 24,083 | ||||||
| Total current assets | 1,664,778 | 2,024,364 | ||||||
| INTANGIBLE ASSETS, net | 427,621 | 434,534 | ||||||
| RIGHT TO USE LEASED ASSETS | 22,015 | - | ||||||
| TOTAL ASSETS | $ | 2,114,414 | $ | 2,458,898 | ||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT | ||||||||
| CURRENT LIABILITIES | ||||||||
| Accrued payroll and payroll related expenses, current portion | $ | 3,471,812 | $ | 3,428,011 | ||||
| Accounts payable and accrued expenses | 1,706,117 | 1,996,097 | ||||||
| Fees payable to directors | 418,546 | 418,546 | ||||||
| Accrued separation costs, current portion | 9,000 | 9,000 | ||||||
| Current portion of related party notes payable | 575,000 | - | ||||||
| Related party convertible note payable | $ | 537,848 | - | |||||
| Convertible notes payable, net of discount | $ | 256,698 | - | |||||
| Employee settlement | 50,000 | 50,000 | ||||||
| Lease liability, current portion | 17,129 | - | ||||||
| Derivative liability on convertible note payable | 246,414 | - | ||||||
| Total current liabilities | 7,288,564 | 5,901,654 | ||||||
| NON-CURRENT LIABILITIES | ||||||||
| Related party notes payable, net of current portion | 1,000,000 | - | ||||||
| Accrued separation costs, less current portion | 85,885 | 92,635 | ||||||
| Lease liability, less current portion | 4,886 | - | ||||||
| Total non-current liabilities | 1,090,771 | 92,635 | ||||||
| COMMITMENTS AND CONTINGENCIES | - | - | ||||||
| Total liabilities | 8,379,335 | 5,994,289 | ||||||
| STOCKHOLDERS’ DEFICIT | ||||||||
| Preferred Stock - $0.001 par value; 50,000,000 shares authorized, 0 shares issued and outstanding as of September 30, 2019 and December 31, 2018, respectively | - | - | ||||||
| Common stock - $0.001 par value; 400,000,000 shares authorized, 137,261,594 and 133,888,573 shares issued and outstanding as of September 30, 2019 and December 31, 2018, respectively | 137,262 | 133,889 | ||||||
| Additional paid-in-capital | 59,191,875 | 58,274,038 | ||||||
| Accumulated deficit | (65,594,058 | ) | (61,943,318 | ) | ||||
| Total stockholders’ deficit | (6,264,921 | ) | (3,535,391 | ) | ||||
| TOTAL LIABILITIES AND STOCKHOLDERS’ DEFICIT | $ | 2,114,414 | $ | 2,458,898 | ||||
The accompanying notes are an integral part of these Condensed Consolidated Financial Statements.
| F-27 |
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS
| For the three-months ended | For the nine-months ended | |||||||||||||||
| September 30, | September 30, | |||||||||||||||
| 2019 | 2018 | 2019 | 2018 | |||||||||||||
| (Unaudited) | (Unaudited) | (Unaudited) | (Unaudited) | |||||||||||||
| REVENUES, net | $ | 229,142 | $ | 549,540 | $ | 439,505 | $ | 1,134,899 | ||||||||
| COST OF GOODS SOLD | 120,818 | 240,152 | 254,479 | 521,353 | ||||||||||||
| GROSS PROFIT | 108,324 | 309,388 | 185,026 | 613,546 | ||||||||||||
| OPERATING EXPENSES: | ||||||||||||||||
| Salaries and wages | 387,636 | 387,119 | 1,177,362 | 1,202,576 | ||||||||||||
| Professional fees | 375,298 | 225,875 | 817,546 | 637,042 | ||||||||||||
| Selling, general, and administrative expenses | 206,042 | 350,630 | 731,487 | 1,168,747 | ||||||||||||
| Stock based compensation | 175,712 | 180,562 | 534,774 | 443,249 | ||||||||||||
| Research and development | 145,273 | 86,115 | 250,141 | 214,093 | ||||||||||||
| Depreciation and amortization | 10,074 | 6,718 | 29,102 | 23,853 | ||||||||||||
| Total operating expenses | 1,300,035 | 1,237,019 | 3,540,412 | 3,689,560 | ||||||||||||
| Loss from operations | (1,191,711 | ) | (927,631 | ) | (3,355,386 | ) | (3,076,014 | ) | ||||||||
| OTHER INCOME (EXPENSE): | ||||||||||||||||
| Interest income | 3 | 7 | 5 | 1,941 | ||||||||||||
| Other income | - | - | - | 556 | ||||||||||||
| Change in fair value of derivative liability | (20,524 | ) | - | (3,139 | ) | - | ||||||||||
| Loss on abandonment of patents | (36,205 | ) | - | (36,205 | ) | - | ||||||||||
| Interest expense | (185,189 | ) | (1,264 | ) | (256,015 | ) | (3,356 | ) | ||||||||
| Total other (expense) income, net | (241,915 | ) | (1,257 | ) | (295,354 | ) | (859 | ) | ||||||||
| Loss before the provision for income taxes | (1,433,626 | ) | (928,888 | ) | (3,650,740 | ) | (3,076,873 | ) | ||||||||
| PROVISION FOR INCOME TAXES | - | - | - | - | ||||||||||||
| NET LOSS | $ | (1,433,626 | ) | $ | (928,888 | ) | $ | (3,650,740 | ) | $ | (3,076,873 | ) | ||||
| NET LOSS PER SHARE | ||||||||||||||||
| Basic | $ | (0.01 | ) | $ | (0.01 | ) | $ | (0.03 | ) | $ | (0.02 | ) | ||||
| Diluted | $ | (0.01 | ) | $ | (0.01 | ) | $ | (0.03 | ) | $ | (0.02 | ) | ||||
| SHARES USED IN CALCULATION OF NET LOSS PER SHARE | ||||||||||||||||
| Basic | 136,640,761 | 130,083,598 | 135,516,490 | 125,271,516 | ||||||||||||
| Diluted | 136,640,761 | 130,083,598 | 135,516,490 | 125,271,516 | ||||||||||||
The accompanying notes are an integral part of these Condensed Consolidated Financial Statements.
| F-28 |
CONDENSED CONSOLIDATED STATEMENT OF CHANGES IN STOCKHOLDER DEFICIT
For the nine-months ended September 30, 2018 and 2019
| Common Stock | Additional Paid-In- | Deferred | Accumulated | |||||||||||||||||||||
| Shares | Amount | Capital | Compensation | Deficit | Total | |||||||||||||||||||
| Balance at January 1, 2018 | 122,674,516 | $ | 122,675 | $ | 56,401,069 | $ | (10,125 | ) | $ | (57,919,096 | ) | $ | (1,405,477 | ) | ||||||||||
| Common stock grants to independent directors | 906,774 | 907 | 199,093 | - | - | 200,000 | ||||||||||||||||||
| Deferred compensation | - | - | - | 10,125 | - | 10,125 | ||||||||||||||||||
| Cardax 2018 Warrant Exchange Offering | 9,600,286 | 9,600 | 1,234,437 | - | - | 1,244,037 | ||||||||||||||||||
| Stock option exercises - cashless | 156,997 | 157 | (157 | ) | - | - | - | |||||||||||||||||
| Stock based compensation - options | - | - | 233,124 | - | - | 233,124 | ||||||||||||||||||
| Net loss | - | - | - | - | (3,076,873 | ) | (3,076,873 | ) | ||||||||||||||||
| Balance at September 30, 2018 | 133,338,573 | $ | 133,339 | $ | 58,067,566 | $ | - | $ | (60,995,969 | ) | $ | (2,795,064 | ) | |||||||||||
| Balance at January 1, 2019 | 133,888,573 | $ | 133,889 | $ | 58,274,038 | $ | - | $ | (61,943,318 | ) | $ | (3,535,391 | ) | |||||||||||
| Common stock grants to independent directors | 1,627,191 | 1,627 | 260,873 | - | - | 262,500 | ||||||||||||||||||
| Common stock grant to service providers | 112,500 | 113 | 14,287 | - | - | 14,400 | ||||||||||||||||||
| Stock based compensation - options | - | - | 257,875 | - | - | 257,875 | ||||||||||||||||||
| Restricted stock issuances | 1,633,330 | 1,633 | 243,367 | - | - | 245,000 | ||||||||||||||||||
| Issuance of warrants attached to a convertible note | - | - | 141,435 | - | - | 141,435 | ||||||||||||||||||
| Net loss | - | - | - | - | (3,650,740 | ) | (3,650,740 | ) | ||||||||||||||||
| Balance at September 30, 2019 | 137,261,594 | $ | 137,262 | $ | 59,191,875 | $ | - | $ | (65,594,058 | ) | $ | (6,264,921 | ) | |||||||||||
The accompanying notes are an integral part of these Condensed Consolidated Financial Statements.
| F-29 |
Cardax, Inc., and Subsidiary
CONDENSED CONSOLIDATED STATEMENT OF CHANGES IN STOCKHOLDER DEFICIT
(continued)
For the three-months ended September 30, 2018 and 2019
| Common Stock | Additional Paid-In- | Deferred | Accumulated | |||||||||||||||||||||
| Shares | Amount | Capital | Compensation | Deficit | Total | |||||||||||||||||||
| Balance at July 1, 2018 | 123,300,787 | $ | 123,301 | $ | 56,653,005 | $ | - | $ | (60,067,081 | ) | $ | (3,290,775 | ) | |||||||||||
| Common stock grants to independent directors | 437,500 | 438 | 87,062 | - | - | 87,500 | ||||||||||||||||||
| Cardax 2018 Warrant Exchange Offering | 9,600,286 | 9,600 | 1,234,437 | - | - | 1,244,037 | ||||||||||||||||||
| Deferred compensation | - | - | - | - | - | - | ||||||||||||||||||
| Stock based compensation - options | - | - | 93,062 | - | - | 93,062 | ||||||||||||||||||
| Net loss | - | - | - | - | (928,888 | ) | (928,888 | ) | ||||||||||||||||
| Balance at September 30, 2018 | 133,338,573 | $ | 133,339 | $ | 58,067,566 | $ | - | $ | (60,995,969 | ) | $ | (2,795,064 | ) | |||||||||||
| Balance at July 1, 2019 | 136,640,761 | $ | 136,641 | $ | 58,908,648 | $ | - | $ | (64,160,432 | ) | $ | (5,115,143 | ) | |||||||||||
| Common stock grants to independent directors | 583,333 | 583 | 86,917 | - | - | 87,500 | ||||||||||||||||||
| Common stock grant to service providers | 37,500 | 38 | 3,300 | - | - | 3,338 | ||||||||||||||||||
| Stock based compensation - options | - | - | 84,875 | - | - | 84,875 | ||||||||||||||||||
| Issuance of warrants attached to a convertible note | - | - | 108,135 | - | - | 108,135 | ||||||||||||||||||
| Net loss | - | - | - | - | (1,433,626 | ) | (1,433,626 | ) | ||||||||||||||||
| Balance at September 30, 2019 | 137,261,594 | $ | 137,262 | $ | 59,191,875 | $ | - | $ | (65,594,058 | ) | $ | (6,264,921 | ) | |||||||||||
The accompanying notes are an integral part of these Condensed Consolidated Financial Statements.
| F-30 |
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
For the nine-months ended September 30, 2018 and 2019
| 2019 | 2018 | |||||||
| (Unaudited) | (Unaudited) | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net loss | $ | (3,650,740 | ) | $ | (3,076,873 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | 29,102 | 23,853 | ||||||
| Amortization of debt discount | 129,256 | - | ||||||
| Stock based compensation | 534,775 | 443,249 | ||||||
| Bad debt expense on note receivable and accrued interest | - | 89,933 | ||||||
| Loss on abandonment of patents | 36,205 | - | ||||||
| Change in fair value of derivative liability | 3,139 | |||||||
| Changes in assets and liabilities: | ||||||||
| Accounts receivable | 32,333 | (193,168 | ) | |||||
| Inventories | 172,653 | 14,251 | ||||||
| Deposits and other assets | - | (118,168 | ) | |||||
| Prepaid expenses | (21,013 | ) | (1,214 | ) | ||||
| Accrued payroll and payroll related expenses | 43,801 | 55,230 | ||||||
| Accounts payable and accrued expenses | (350,650 | ) | 50,752 | |||||
| Accrued separation costs | (6,750 | ) | - | |||||
| Net cash used in operating activities | (3,047,889 | ) | (2,712,155 | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||
| Increase in intangible assets | (58,394 | ) | (30,483 | ) | ||||
| Net cash used in investing activities | (58,394 | ) | (30,483 | ) | ||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Proceeds from the issuances of related party notes payable | 1,575,000 | - | ||||||
| Proceeds from the issuance of a related party convertible note payable | 750,000 | - | ||||||
| Proceeds from the issuances of convertible notes payable | 300,000 | - | ||||||
| Proceeds from the issuance of common stock | 245,000 | 704,375 | ||||||
| Net cash provided by financing activities | 2,870,000 | 704,375 | ||||||
| NET DECREASE IN CASH | (236,283 | ) | (2,038,263 | ) | ||||
| BEGINNING OF THE PERIOD | 243,753 | 2,236,837 | ||||||
| END OF THE PERIOD | $ | 7,470 | $ | 198,574 | ||||
| SUPPLEMENTAL DISCLOSURES: | ||||||||
| Cash paid for interest | $ | 13,937 | $ | 3,356 | ||||
| Cash paid for income taxes | $ | - | $ | - | ||||
| NON-CASH INVESTING AND FINANCING ACTIVITIES: | ||||||||
| Discount recognized on notes payable at issuance | $ | 384,710 | $ | - | ||||
| Settlement of receivables with payables | $ | 60,670 | $ | 221,814 | ||||
| Right to use assets funded through leases | $ | 22,015 | $ | 539,662 | ||||
The accompanying notes are an integral part of these Condensed Consolidated Financial Statements.
| F-31 |
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS
NOTE 1 – COMPANY BACKGROUND
The Company’s predecessor, Cardax Pharmaceuticals, Inc. (“Holdings”), was incorporated in the State of Delaware on March 23, 2006.
Cardax, Inc. (the “Company”) (OTCQB:CDXI) is a development stage biopharmaceutical company primarily focused on the development of pharmaceuticals for chronic diseases driven by inflammation. The Company also has a commercial business unit that markets dietary supplements for inflammatory health. CDX-101, the Company’s astaxanthin pharmaceutical candidate, is being developed for cardiovascular inflammation and dyslipidemia, with a target initial indication of severe hypertriglyceridemia. CDX-301, the Company’s zeaxanthin pharmaceutical candidate, is being developed for macular degeneration, with a target initial indication of Stargardt disease. The Company’s pharmaceutical candidates are currently in pre-clinical development, including the planning of IND enabling studies. ZanthoSyn® is a physician recommended astaxanthin dietary supplement for inflammatory health. The Company sells ZanthoSyn® primarily through wholesale and e-commerce channels. The safety and efficacy of the Company’s products have not been directly evaluated in clinical trials or confirmed by the FDA.
Going concern matters
The accompanying condensed consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. As shown in the accompanying condensed consolidated financial statements, the Company incurred net losses of $1,433,626 and $3,650,740 for the three and nine-months ended September 30, 2019, respectively, and incurred net losses of $928,888 and $3,076,873 for the three and nine-months ended September 30, 2018, respectively. The Company has incurred losses since inception resulting in an accumulated deficit of $65,594,058 as of September 30, 2019, and has had negative cash flows from operating activities since inception. The Company expects that its marketing program for ZanthoSyn® will continue to focus on outreach to physicians, healthcare professionals, retail personnel, and consumers, and anticipates further losses in the development of its consumer business. The Company also plans to advance the research and development of its pharmaceutical candidates and anticipates further losses in the development of its pharmaceutical business. As a result of these and other factors, management has determined there is substantial doubt about the Company’s ability to continue as a going concern.
During the nine-months ended September 30, 2019, the Company raised additional capital to carry out its business plan. As part of the Company’s efforts, it raised an additional $245,000 in equity from existing stockholders and $2,625,000 in gross proceeds from debt, including $2,325,000 from related parties. On August 14, 2019, the Company filed a registration statement on Form S-1 for a proposed $15 million public offering of common stock and warrants. The Company intends to use the proceeds from the proposed public offering primarily to fund pharmaceutical development and its operations. The Company’s continued ability to raise additional capital through future equity and debt securities issuances is unknown. Obtaining additional financing, the successful development of the Company’s contemplated plan of operations, and its transition, ultimately, to profitable operations are necessary for the Company to continue operations. The ability to successfully resolve these factors raises substantial doubt about the Company’s ability to continue as a going concern. The condensed consolidated financial statements of the Company do not include any adjustments that may result from the outcome of these uncertainties.
| F-32 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Unaudited interim financial information
The accompanying unaudited condensed consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) for interim financial information and the rules and regulations of the Securities and Exchange Commission (the “SEC”) for interim financial information. In the opinion of the Company’s management, the accompanying condensed consolidated financial statements reflect all adjustments, consisting of normal, recurring adjustments, considered necessary for a fair presentation of the results for the interim periods ended September 30, 2019 and 2018.
Although management believes that the disclosures in these unaudited condensed consolidated financial statements are adequate to make the information presented not misleading, certain information and footnote disclosures normally included in financial statements that have been prepared in accordance U.S. GAAP have been condensed or omitted pursuant to the rules and regulations of the SEC.
These unaudited interim consolidated financial statements should be read in conjunction with the consolidated financial statements and the related notes included in the Company’s annual report on Form 10-K for the fiscal year ended December 31, 2018, filed with the SEC on March 28, 2019.
Revenue from contracts with customers
In May 2014, the Financial Accounting Standards Board (“FASB”) issued a new standard related to revenue recognition. Under the standard, revenue is recognized when a customer obtains control of promised goods or services in an amount that reflects the consideration the entity expects to receive in exchange for those goods or services. In addition, the standard requires disclosure of the nature, amount, timing, and uncertainty of revenue and cash flows arising from contracts with customers.
The Company adopted this standard effective January 1, 2018, using the retrospective method. As there was no impact on contracts that were previously completed and no significant impact to contracts completed after adoption, there was no need to restate prior results from operations.
The Company recognizes revenues from its contracts with customers for its products through wholesale and e-commerce channels when goods and services have been identified, the payment terms agreed to, the contract has commercial substance, both parties have approved the contract, and it is probable that the Company will collect all substantial consideration.
The following table presents our revenues disaggregated by revenue source and geographical location. Sales and usage-based taxes are included as a component of revenues for the nine-months ended:
| September 30, 2019 | September 30, 2018 | |||||||||
| Geographical area | Source | (Unaudited) | (Unaudited) | |||||||
| United States | Nutraceuticals | $ | 439,505 | $ | 1,118,486 | |||||
| Hong Kong | Nutraceuticals | $ | - | $ | 16,413 | |||||
| F-33 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
Revenue from contracts with customers (continued)
Sales discounts, rebates, promotional amounts to vendors, and returns and allowances are recorded as a reduction to sales in the period in which sales are recorded. The Company records shipping charges and sales tax gross in revenues and cost of goods sold. Sales discounts and other adjustments are recorded at the time of sale.
Leases
In February 2016, the FASB issued ASU No. 2016-02, Leases. This ASU requires management to recognize lease assets and lease liabilities for all leases. ASU No. 2016-02 retains a distinction between finance leases and operating leases. The classification criteria for distinguishing between finance leases and operating leases are substantially similar to the classification criteria for distinguishing between capital leases and operating leases in the previous lease guidance. The result of retaining a distinction between finance leases and operating leases is that under the lessee accounting model, the effect of leases in the statement of comprehensive income and the statement of cash flows is largely unchanged from previous U.S. GAAP. The guidance in ASU No. 2016-02 is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years.
The Company applied the modified retrospective approach in adopting this standard. The modified retrospective approach includes a number of optional practical expedients that the Company elected to apply; primarily the identification and classification of leases that commenced before the effective date, initial direct costs for leases that commenced before the effective date, and the ability to use hindsight in evaluating lessee options to extend or terminate a lease or to purchase the underlying asset. As part of this adoption, the Company will, in effect, continue to account for leases that commence before the effective date in accordance with previous U.S. GAAP unless the lease is modified, except that lessees are required to recognize a right-of-use asset and a lease liability for all operating leases at each reporting date based on the present value of the remaining minimum rental payments that were tracked and disclosed under previous U.S. GAAP. This adoption of this standard on January 1, 2019, resulted in the Company recognizing a right-to-use asset and lease liability. The Company elected to not recognize any right-to-use assets or liabilities for leases that are twelve months or less. Lease costs are recognized straight-line over the term of the lease. The adoption of this standard did not impact retained earnings or cash flows of the Company.
Derivative financial instruments
The Company accounts for the fair value of the conversion feature in accordance with ASC 815-15, Derivatives and Hedging; Embedded Derivatives, which requires the Company to bifurcate and separately account for the conversion feature as an embedded derivative contained in the Company’s convertible note. The Company is required to carry the embedded derivative on its balance sheet at fair value. The initial value of the embedded derivative is accounted for as a discount to the convertible note and a derivative liability. The liability is required to be remeasured at each reporting date and changes in fair value is recognized as a component in its results of operations. The Company valued the embedded derivatives on the condensed consolidated balance sheet at fair value using the Black-Scholes valuation model.
| F-34 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
Other significant accounting policies
There have been no other material changes to our significant accounting policies during the nine-months ended September 30, 2019, as compared to the significant accounting policies described in our Annual Report.
Reclassifications
The Company has made certain reclassifications to conform its prior periods’ data to the current presentation, such as reclassifying a separation agreement that has terms extending beyond one year. These reclassifications had no effect on the reported results of operations or cash flows.
NOTE 3 – INVENTORIES
Inventories consist of the following as of:
| September
30, 2019 (Unaudited) | December 31, 2018 | |||||||
| Finished goods | $ | 533,139 | $ | 96,750 | ||||
| Raw materials | 774,588 | 1,383,630 | ||||||
| Total inventories | $ | 1,307,727 | $ | 1,480,380 | ||||
As of September 30, 2019 and December 31, 2018, all raw materials were held at the manufacturer’s facility for future production.
NOTE 4 – INTANGIBLE ASSETS, net
Intangible assets, net, consists of the following as of:
| September 30, 2019 (Unaudited) | December 31, 2018 | |||||||
| Patents | $ | 613,943 | $ | 578,326 | ||||
| Less accumulated amortization | (321,614 | ) | (292,512 | ) | ||||
| 292,329 | 285,814 | |||||||
| Patents pending | 135,292 | 148,720 | ||||||
| Total intangible assets, net | $ | 427,621 | $ | 434,534 | ||||
Patents are amortized straight-line over a period of fifteen years. Amortization expense was $10,074 and $29,102 for the three and nine-months ended September 30, 2019, respectively. Amortization expense was $6,717 and $21,952 for the three and nine-months ended September 30, 2018, respectively.
| F-35 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 4 – INTANGIBLE ASSETS, net (continued)
The Company has capitalized costs for several patents that are still pending. In those instances, the Company has not recorded any amortization. The Company will commence amortization when these patents are approved.
During the three and nine-months ended September 30, 2019, the Company abandoned three patent applications in progress resulting in a loss of $36,205 on the abandonment of patents.
The Company owns 29 issued patents, including 14 in the United States and 15 others in Europe, Canada, China, India, Japan, and Hong Kong. These patents will expire beginning in 2023 through 2028, subject to any patent term extensions of the individual patent. The Company has 2 patent applications pending in the United States and 2 foreign patent applications pending in Europe and the Patent Cooperation Treaty (“PCT”) countries.
NOTE 5 –ACCRUED SEPARATION COSTS
On August 9, 2016, the Company entered into a separation agreement with an employee to pay $118,635 of accrued compensation over nine-years. As of September 30, 2019, $94,885 remains outstanding of which $9,000 is due within one-year and is reflected as a current liability.
| F-36 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 6 – RELATED PARTY NOTES PAYABLE
Notes payable consisted of the following as of:
| September 30, 2019 | December 31, 2018 | |||||||
| (Unaudited) | ||||||||
| Inventory financing. On January 11, 2019, the Company entered into a $1,000,000 revolving inventory financing facility with a lender that is also a current stockholder that beneficially owns more than 5% of the Company’s common stock. Use of proceeds from this facility is limited to the purchase of inventory, including raw materials, intermediates, and finished goods, unless otherwise waived by the lender. This facility accrues interest at the rate of 12% per annum, is unsecured, and matures in three years from origination. This facility requires monthly interest payments. | $ | 1,000,000 | $ | - | ||||
| Officer loan. On June 26, 2019, the Company borrowed $75,000 from the Chief Executive Officer of the Company with principal and interest due on August 26, 2019, which was subsequently extended to December 31, 2019. This note accrues interest at the rate of 4.5% per annum and is unsecured. | 75,000 | |||||||
| Promissory note. On May 20, 2019, the Company entered into a $400,000 promissory note with a lender that is also a current stockholder that beneficially owns more than 5% of the Company’s common stock. On July 10, 2019, this note was amended to increase the principal sum by an additional $100,000. This note accrues interest at the rate of 12% per annum, is unsecured, and originally matured on August 20, 2019, which was subsequently extended to June 30, 2020. All principal and accrued interest is due on the maturity date. | 500,000 | - | ||||||
| Total notes payable | $ | 1,575,000 | $ | - | ||||
| Less current portion | (575,000 | ) | - | |||||
| Long term notes payable | $ | 1,000,000 | $ | - | ||||
Interest expense
The Company incurred interest charges of $45,925 and $101,385 during the three and nine-months ended September 30, 2019, respectively, on these notes payable of which $31,111 was accrued and payable as of September 30, 2019.
| F-37 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 6 – RELATED PARTY NOTES PAYABLE (continued)
Maturities
Future maturities of notes payable are as follows as of September 30:
| 2020 | $ | 575,000 | ||
| 2021 | - | |||
| 2022 | 1,000,000 | |||
| $ | 1,575,000 |
NOTE 7 – RELATED PARTY CONVERTIBLE NOTE PAYABLE
Related party convertible note payable consisted of the following as of:
| September 30, 2019 | December 31, 2018 | |||||||
| (Unaudited) | ||||||||
| Convertible note 2019-02. On July 19, 2019, the Company issued a convertible note payable in the amount $815,217, with an original issue discount of $65,217 in exchange for $750,000. This note accrues interest at 8% per annum and matures on June 30, 2020. This note and accrued interest may convert into shares of common stock at the conversion price then in effect (initially $0.12 per share, subject to adjustment) any time at the holder’s option or automatically upon a qualified financing of at least $5 million at the lower of the conversion price then in effect or a 25% discount to the offering price. The conversion price is subject to adjustment upon the issuance of the Company’s common stock or securities convertible into common stock at a price per share less than the then prevailing conversion price, other than specified exempt issuances; accordingly, on November 8, 2019, the conversion price was adjusted to $0.07 per share. This note was also issued with a detachable warrant to purchase 1,500,000 shares of stock at $0.12 per share, which shall be adjusted in accordance with any adjustment to the conversion price of this note; accordingly, on November 8, 2019, the exercise price was adjusted to $0.07 per share. The valuation of the conversion feature and detachable warrants resulted in the recognition of an additional $286,050 discount on this note. This note requires monthly interest payments. | $ | 815,217 | $ | - | ||||
| Total notes payable | 815,217 | - | ||||||
| Less original issue discounts | (65,217 | ) | - | |||||
| Related party convertible note payable, net | 750,000 | - | ||||||
| Less conversion rights and warrant discounts | (286,050 | ) | - | |||||
| Plus amortization of discounts | 73,898 | - | ||||||
| Total convertible notes payable, net | $ | 537,848 | $ | - | ||||
| F-38 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 7 – RELATED PARTY CONVERTIBLE NOTE PAYABLE (continued)
Discounts
Total discounts of $351,267 are amortized using the interest method, which resulted in amortization recorded as interest expense of $73,898 for the three and nine-months ended September 30, 2019.
Interest expense
The Company incurred interest charges of $13,222 during the three and nine-months ended September 30, 2019, on this related party convertible note payable of which $5,360 was accrued and payable as of September 30, 2019.
Maturities
Future maturities of notes payable are as follows as of September 30:
| 2020 | $ | 815,217 | ||
| $ | 815,217 |
| F-39 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 8 – CONVERTIBLE NOTES PAYABLE
Convertible notes payable consisted of the following as of:
| September 30, 2019 | December 31, 2018 | |||||||
| (Unaudited) | ||||||||
| Convertible note 2019-01. On April 18, 2019, the Company issued a convertible note payable in the amount $150,000. This note accrues interest at 10% per annum and matures on December 31, 2019. This note and accrued interest may convert into shares of common stock at the conversion price then in effect (initially $0.12 per share, subject to adjustment) any time at the holder’s option or automatically upon maturity provided the 20-day volume weighted average price per share of the Company’s common stock upon maturity is at least $0.12 per share. The conversion price is subject to adjustment upon the issuance of the Company’s common stock or securities convertible into common stock at a price per share less than the then prevailing conversion price, other than specified exempt issuances; accordingly, on November 8, 2019, the conversion price was adjusted to $0.07 per share. This note was also issued with a detachable warrant to purchase 500,000 shares of stock at $0.20 per share. The valuation of the conversion feature and detachable warrants resulted in the recognition of an $83,300 aggregate discount on this note. | $ | 150,000 | $ | - | ||||
| Convertible note 2019-03. On September 4, 2019, the Company issued a convertible note payable in the amount $108,696, with an original issue discount of $8,696 in exchange for $100,000. This note accrues interest at 8% per annum and matures on June 30, 2020. This note and accrued interest may convert into shares of common stock at $0.12 per share any time at the holder’s option. If this note, or any portion thereof, has not been repaid or converted in full on or prior to the maturity date, then repayment of the unpaid principal balance plus any accrued and unpaid interest thereon, shall be amortized over the following thirty-six (36) months. This note was also issued with a detachable warrant to purchase 200,000 shares of stock at $0.12 per share. The valuation of the detachable warrants resulted in the recognition of an additional $11,170 discount on this note. This note requires monthly interest payments. | 108,696 | - | ||||||
| F-40 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 8 – CONVERTIBLE NOTES PAYABLE (continued)
| September 30, 2019 | December 31, 2018 | |||||||
| (Unaudited) | ||||||||
| Convertible note 2019-04. On September 25, 2019, the Company issued a convertible note payable in the amount $54,348, with an original issue discount of $4,348 in exchange for $50,000. This note accrues interest at 8% per annum and matures on June 30, 2020. This note and accrued interest may convert into shares of common stock at $0.12 per share any time at the holder’s option. If this note, or any portion thereof, has not been repaid or converted in full on or prior to the maturity date, then repayment of the unpaid principal balance plus any accrued and unpaid interest thereon, shall be amortized over the following thirty-six (36) months. This note was also issued with a detachable warrant to purchase 100,000 shares of stock at $0.12 per share. The valuation of the detachable warrants resulted in the recognition of an additional $4,190 discount on this note. This note requires monthly interest payments. | 54,348 | - | ||||||
| Total notes payable | 313,044 | - | ||||||
| Less original issue discounts | (13,044 | ) | - | |||||
| Convertible notes payable, net | 300,000 | - | ||||||
| Less conversion rights and warrant discounts | (98,660 | ) | - | |||||
| Plus amortization of discounts | 55,358 | - | ||||||
| Total convertible notes payable, net | $ | 256,698 | $ | - | ||||
Discounts
Total discounts of $111,704 are amortized using the interest method, which resulted in amortization recorded as interest expense of $31,696 and $55,358 for the three and nine-months ended September 30, 2019, respectively.
Interest expense
The Company incurred interest charges of $4,496 and $7,537 during the three and nine-months ended September 30, 2019, respectively, on these notes payable of which $7,537 was accrued and payable as of September 30, 2019.
Maturities
Future maturities of notes payable are as follows as of September 30:
| 2020 | $ | 313,044 | ||
| $ | 313,044 |
| F-41 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 9 – DERIVATIVE FINANCIAL INSTRUMENTS
The Company has identified the embedded derivatives related to the convertible notes described in Note 8. These embedded derivatives included certain conversion and reset features. The accounting treatment of derivative financial instruments requires that the Company record fair value of these derivative liabilities as of the inception date of those convertible notes and each subsequent reporting date.
The Company estimates the fair value of these derivative liabilities using the Black-Scholes valuation model. The initial value is used in the determination of a note discount with each subsequent change in fair value as a component of operations. The range of fair value assumptions used for derivative financial instruments during the nine-months ended September 30, 2019, were as follows:
| Dividend yield | 0.0 | % | ||
| Risk-free rate | 1.75% - 2.44 | % | ||
| Volatility | 102% - 137 | % | ||
| Expected term | 1 year |
Volatility was calculated based on the historical volatility of the Company. The risk-free interest rate used was based on the U.S. Treasury constant maturity rate in effect at the time of grant for the expected term of the derivative liabilities to be valued. The expected dividend yield was zero, because the Company does not anticipate paying a dividend within the relevant timeframe.
For the nine-months ended September 30, 2019, the Company recognized total derivative liabilities and convertible note discounts of $243,275 based on the fair value at the convertible notes’ inception dates. These derivative liabilities were subsequently revalued at $246,414 as of September 30, 2019, which resulted in a loss of $3,139 on the change in value of these derivative liabilities.
The following table presents the three-level hierarchy prescribed by U.S. GAAP for derivative liabilities since it is a liability that is measured and recognized at fair value on a recurring basis as of:
| Level 1 | Level 2 | Level 3 | ||||||||||
| September 30, 2019 | - | - | $ | 246,414 | ||||||||
NOTE 10 – STOCKHOLDERS’ DEFICIT
Self-directed stock issuance 2019
During the nine-months ended September 30, 2019, the Company sold securities in a self-directed offering to existing stockholders of the Company in the aggregate amount of $245,000, respectively, at $0.30 per unit. Each $0.30 unit consisted of 2 shares of restricted common stock (1,633,330 shares) and a five-year warrant to purchase 1 share of restricted common stock (816,665 warrant shares) at $0.20 per share.
| F-42 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 10 – STOCKHOLDERS’ DEFICIT (continued)
Warrant exchange offering
In June 2018, the Company commenced an offering to exchange outstanding warrants for shares of common stock under a Form S-4 Registration Statement. These shares of common stock were issued to warrant holders in exchange for (i) their outstanding warrants to purchase shares of common stock at $0.625 per share, and (ii) cash payment of $0.15 per share. This offering closed on July 27, 2018, and resulted in an exchange of 9.6 million warrants and $1,440,043 in gross proceeds for 9,600,286 shares of common stock. Stock issuance costs associated with this capital raise totaled $196,006, resulting in a net total of $1,244,037 raised in this offering.
Shares outstanding
As of September 30, 2019 and December 31, 2018, the Company had a total of 137,261,594 and 133,888,573, respectively, shares of common stock outstanding.
NOTE 11 – STOCK GRANTS
Director stock grants
During the nine-months ended September 30, 2019 and 2018, the Company granted its independent directors an aggregate of 1,627,191 and 906,774, respectively, shares of restricted common stock in the Company. These shares were fully vested upon issuance. The increase in number of shares issued was due to the expansion of the Board of Directors by two members in September 2018. The expense recognized for these grants based on the grant date fair value was $262,500 and $200,000 for the nine-months ended September 30, 2019 and 2018, respectively.
Consultant stock grants
On April 10, 2017, the Company granted a consultant 100,000 shares of restricted common stock valued at $0.23 per share. These shares were subject to a risk of forfeiture and vested quarterly in arrears commencing on April 1, 2017. The Company recognized $0 and $5,750 in stock-based compensation related to this grant during the nine-months ended September 30, 2019 and 2018, respectively.
On August 8, 2017, the Company granted a consultant 100,000 shares of restricted common stock valued at $0.175 per share. These shares were subject to a risk of forfeiture and vested 25% upon grant and quarterly in arrears thereafter commencing on September 1, 2017. The Company recognized $0 and $4,375 in stock-based compensation related to this grant during the nine-months ended September 30, 2019 and 2018, respectively.
On December 31, 2018, the Company granted consultants 112,500 shares of restricted common stock valued at $0.20 per share. These shares were fully vested upon issuance. The Company recognized $22,500 in stock-based compensation related to these grants during the year ended December 31, 2018.
On March 31, 2019, the Company granted consultants 37,500 shares of restricted common stock valued at $0.17 per share. On June 30, 2019, the Company granted consultants 37,500 shares of restricted common stock valued at $0.125 per share. On September 30, 2019, the Company granted consultants 37,500 shares of restricted common stock valued at $0.089 per share. These shares were fully vested upon issuance. The Company recognized $14,400 in stock-based compensation related to these grants during the nine-months ended September 30, 2019.
| F-43 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 12 – STOCK OPTION PLANS
On February 7, 2014, the Company adopted the 2014 Equity Compensation Plan. Under this plan, the Company may issue options to purchase shares of common stock to employees, directors, advisors, and consultants. The aggregate number of shares reserved under this plan upon adoption was 30,420,148. On April 16, 2015, the majority stockholder of the Company approved an increase in the Company’s 2014 Equity Compensation Plan by 15 million shares. On December 4, 2018, the stockholders of the Company approved an increase in the Company’s 2014 Equity Compensation Plan by an additional 5 million shares, for a total of 50,420,148 shares reserved under the plan.
Under the terms of the 2014 Equity Compensation Plan and the 2006 Stock Incentive Plan (collectively, the “Plans”), incentive stock options may be granted to employees at a price per share not less than 100% of the fair market value at date of grant. If the incentive stock option is granted to a 10% stockholder, then the purchase or exercise price per share shall not be less than 110% of the fair market value per share of common stock on the grant date. Non-statutory stock options and restricted stock may be granted to employees, directors, advisors, and consultants at a price per share, not less than 100% of the fair market value at date of grant. Options granted are exercisable, unless specified differently in the grant documents, over a default term of ten years from the date of grant and generally vest over a period of four years.
A summary of stock option activity is as follows:
| Options | Weighted average exercise price |
Weighted average remaining contractual term in years |
Aggregate intrinsic value |
|||||||||||||
| Outstanding January 1, 2018 | 38,213,427 | $ | 0.41 | 5.23 | $ | 562,456 | ||||||||||
| Exercisable January 1, 2018 | 36,213,427 | $ | 0.41 | 4.98 | $ | 562,456 | ||||||||||
| Canceled | (350,000 | ) | ||||||||||||||
| Granted | 2,833,334 | |||||||||||||||
| Exercised | (200,000 | ) | ||||||||||||||
| Forfeited | - | |||||||||||||||
| Outstanding December 31, 2018 | 40,496,761 | $ | 0.40 | 4.52 | $ | 986,808 | ||||||||||
| Exercisable December 31, 2018 | 37,157,179 | $ | 0.41 | 4.10 | $ | 966,808 | ||||||||||
| Canceled | (58,336 |
) | ||||||||||||||
| Granted | - | |||||||||||||||
| Exercised | - | |||||||||||||||
| Forfeited | - | |||||||||||||||
| Outstanding September 30, 2019 | 40,438,425 | $ | 0.40 | 3.77 | $ | 149,089 | ||||||||||
| Exercisable September 30, 2019 | 38,130,093 | $ | 0.41 | 3.48 | $ | 149,089 | ||||||||||
| F-44 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 12 – STOCK OPTION PLANS (continued)
The aggregate intrinsic value in the table above is before applicable income taxes and represents the excess amount over the exercise price option recipients would have received if all options had been exercised on September 30, 2019, based on a valuation of the Company’s stock for that day.
A summary of the Company’s non-vested options for the nine-months ended September 30, 2019 and year ended December 31, 2018, are presented below:
| Non-vested at January 1, 2018 | 2,000,000 | |||
| Granted | 2,833,334 | |||
| Vested | (1,143,752 | ) | ||
| Canceled | (350,000 | ) | ||
| Non-vested at December 31, 2018 | 3,339,582 | |||
| Granted | - | |||
| Vested | (972,914 | ) | ||
| Canceled | (58,336 | ) | ||
| Non-vested at September 30, 2019 | 2,308,332 |
The Company estimates the fair value of stock options granted on each grant date using the Black-Scholes option valuation model and recognizes an expense ratably over the requisite service period. The range of fair value assumptions related to options issued were as follows for the:
| Nine-months
ended September 30, 2019 | Year
ended December 31, 2018 | |||||||
| Dividend yield | 0.0 | % | 0.0 | % | ||||
| Risk-free rate | 2.38% - 3.04 | % | 2.38% - 3.04 | % | ||||
| Volatility | 214% - 226 | % | 214% - 226 | % | ||||
| Expected term | 3 - 7 years | 3 - 7 years | ||||||
Volatility was calculated based on the historical volatility of the Company. The risk-free interest rate used was based on the U.S. Treasury constant maturity rate in effect at the time of grant for the expected term of the stock options to be valued. The expected dividend yield was zero, because the Company does not anticipate paying a dividend within the relevant timeframe.
The Company records forfeitures as they occur and reverses compensation cost previously recognized, in the period the award is forfeited, for an award that is forfeited before completion of the requisite service period.
Stock option exercise
During the year ended December 31, 2018, the Company issued 156,997 shares of common stock in connection with the cashless exercise of stock options for 100,000, 50,000, and 50,000 shares of common stock exercisable at $0.06 per share with 43,003 shares of common stock withheld with an aggregate fair market value equal to the aggregate exercise price.
| F-45 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 12 – STOCK OPTION PLANS (continued)
Stock based compensation
The Company recognized stock-based compensation expense related to options during the:
| Nine-months ended September 30 | ||||||||
| 2019 | 2018 | |||||||
| Amount | Amount | |||||||
| Service provider compensation | $ | 133,125 | $ | 76,250 | ||||
| Employee compensation | 124,750 | 156,875 | ||||||
| Total | $ | 257,875 | $ | 233,125 | ||||
NOTE 13 – WARRANTS
The following is a summary of the Company’s warrant activity:
| Warrants | Weighted average exercise price | Weighted average remaining contractual term in years | Aggregate intrinsic value | |||||||||||||
| Outstanding January 1, 2018 | 127,434,122 | $ | 0.24 | 3.15 | $ | 3,957,689 | ||||||||||
| Exercisable January 1, 2018 | 127,434,122 | $ | 0.24 | 3.15 | $ | 3,957,689 | ||||||||||
| Canceled | - | |||||||||||||||
| Granted | 315,010 | |||||||||||||||
| Exercised | (9,600,286 | ) | ||||||||||||||
| Expired | (101,984 | ) | ||||||||||||||
| Outstanding December 31, 2018 | 118,046,862 | $ | 0.20 | 2.32 | $ | 7,848,637 | ||||||||||
| Exercisable December 31, 2018 | 118,046,862 | $ | 0.20 | 2.32 | $ | 7,848,637 | ||||||||||
| Canceled | - | |||||||||||||||
| Granted | 3,116,665 | |||||||||||||||
| Exercised | - | |||||||||||||||
| Expired | (18,405,496 | ) | ||||||||||||||
| Outstanding September 30, 2019 | 102,758,031 | $ | 0.13 | 2.07 | $ | 146,779 | ||||||||||
| Exercisable September 30, 2019 | 102,758,031 | $ | 0.13 | 2.07 | $ | 146,779 | ||||||||||
| F-46 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 13 – WARRANTS (continued)
The Company estimates the fair value of warrants granted on each grant date using the Black-Scholes option valuation model. Volatility is calculated based on the historical volatility of the Company. The risk-free interest rate used is based on the U.S. Treasury constant maturity rate in effect at the time of grant for the expected term of the warrants to be valued. The expected dividend yield is zero, because the Company does not anticipate paying a dividend within the relevant timeframe.
The Company did not recognize any stock-based compensation expense related to warrants during the three-months ended September 30, 2019 and 2018.
Convertible note warrants
Warrants to purchase 2,300,000 shares of common stock at $0.12 to $0.20 per share were issued in connection with the issuance of convertible notes. These warrants were immediately vested and expire in five years and were recorded as discounts on the convertible notes in the aggregate amount of $141,435.
Warrant exchange offering
In June 2018, the Company commenced an offering to exchange outstanding warrants for shares of common stock under a Form S-4 Registration Statement. These shares of common stock were issued to warrant holders in exchange for (i) their outstanding warrants to purchase shares of common stock at $0.625 per share, and (ii) cash payment of $0.15 per share. This offering closed on July 27, 2018, and resulted in an exchange of 9.6 million warrants and $1,440,043 in gross proceeds for 9,600,286 shares of common stock. Stock issuance costs associated with this capital raise totaled $196,006, resulting in a net total of $1,244,037 raised in this offering. As part of this offering, warrants to purchase 315,010 shares of common stock at $0.21 per share were issued to investment bankers for their services.
Warrant expiration
During the nine-months ended September 30, 2019, warrants to purchase an aggregate of 18,405,496 shares of common stock expired. During the year ended December 31, 2018, warrants to purchase an aggregate of 101,984 shares of common stock expired.
| F-47 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 14 – INCOME TAXES
The Company accounts for income taxes using the asset and liability method. Under this method, deferred income tax assets and liabilities are determined based upon the difference between the financial statement carrying amounts and the tax basis of assets and liabilities and are measured using the enacted tax rate expected to apply to taxable income in the years in which the differences are expected to be reversed.
The effective tax rate for the three and three-months ended September 30, 2019 and 2018, differs from the statutory rate of 21% as a result of state taxes (net of Federal benefit), permanent differences, and a reserve against deferred tax assets.
The Company’s valuation allowance was primarily related to the operating losses. The valuation allowance is determined in accordance with the provisions of ASC No. 740, Income Taxes, which requires an assessment of both negative and positive evidence when measuring the need for a valuation allowance. Based on the available objective evidence and the Company’s history of losses, management provides no assurance that the net deferred tax assets will be realized. As of September 30, 2019, and December 31, 2018, the Company has applied a valuation allowance against its deferred tax assets net of the expected income from the reversal of the deferred tax liabilities.
Recent tax legislation
On March 22, 2018, the Tax Cuts and Jobs Act (“TCJA”) was enacted into law, which significantly changes existing U.S. tax law and includes numerous provisions that affect our business, such as reducing the U.S. federal statutory tax rate from 35% to 21% effective January 1, 2018.
Uncertain tax positions
The Company is subject to taxation in the United States and three state jurisdictions. The preparation of tax returns requires management to interpret the applicable tax laws and regulations in effect in such jurisdictions, which could affect the amount of tax paid by the Company. Management, in consultation with its tax advisors, files its tax returns based on interpretations that are believed to be reasonable under the circumstances. The income tax returns, however, are subject to routine reviews by the various taxing authorities. As part of these reviews, a taxing authority may disagree with respect to the tax positions taken by management (“uncertain tax positions”) and therefore may require the Company to pay additional taxes.
Management evaluates the requirement for additional tax accruals, including interest and penalties, which the Company could incur as a result of the ultimate resolution of its uncertain tax positions. Management reviews and updates the accrual for uncertain tax positions as more definitive information becomes available from taxing authorities, completion of tax audits, expiration of statute of limitations, or upon occurrence of other events.
| F-48 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 14 – INCOME TAXES (continued)
Uncertain tax positions (continued)
As of September 30, 2019 and December 31, 2018, there was no liability for income tax associated with unrecognized tax benefits. The Company recognizes accrued interest related to unrecognized tax benefits as well as any related penalties in interest income or expense in its condensed consolidated statements of operations, which is consistent with the recognition of these items in prior reporting periods.
The federal and state income tax returns of the Company are subject to examination by the IRS and state taxing authorities, generally for three years after they were filed.
NOTE 15 – BASIC AND DILUTED NET LOSS PER SHARE
The following table sets forth the computation of the Company’s basic and diluted net loss per share for:
| Three-months ended September 30, 2019 (Unaudited) | ||||||||||||
| Net Loss (Numerator) | Shares (Denominator) | Per
share amount | ||||||||||
| Basic loss per share | $ | (1,433,626 | ) | 136,640,761 | $ | (0.01 | ) | |||||
| Effect of dilutive securities—Common stock options, warrants, and convertible note | - | - | - | |||||||||
| Diluted loss per share | $ | (1,433,626 | ) | 136,640,761 | $ | (0.01 | ) | |||||
| Three-months ended September 30, 2018 (Unaudited) | ||||||||||||
| Net Loss (Numerator) | Shares (Denominator) | Per
share amount | ||||||||||
| Basic loss per share | $ | (928,888 | ) | 130,083,598 | $ | (0.01 | ) | |||||
| Effect of dilutive securities—Common stock options, warrants, and convertible note | - | - | - | |||||||||
| Diluted loss per share | $ | (928,888 | ) | 130,083,598 | $ | (0.01 | ) | |||||
| Nine-months ended September 30, 2019 (Unaudited) | ||||||||||||
| Net Loss (Numerator) | Shares (Denominator) | Per
share amount | ||||||||||
| Basic loss per share | $ | (3,650,740 | ) | 135,516,490 | (0.03 | ) | ||||||
| Effect of dilutive securities—Common stock options, warrants, and convertible note | - | - | - | |||||||||
| Diluted loss per share | $ | (3,650,740 | ) | 135,516,490 | (0.03 | ) | ||||||
| F-49 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 15 – BASIC AND DILUTED NET LOSS PER SHARE (continued)
| Nine-months ended September 30, 2018 (Unaudited) | ||||||||||||
| Net Loss (Numerator) | Shares (Denominator) | Per
share amount | ||||||||||
| Basic loss per share | $ | (3,076,873 | ) | 125,271,516 | $ | (0.02 | ) | |||||
| Effect of dilutive securities—Common stock options, warrants, and convertible note | - | - | - | |||||||||
| Diluted loss per share | $ | 3,076,873 | 125,271,516 | $ | (0.02 | ) | ||||||
The following outstanding shares of common stock equivalents were excluded from the computation of diluted net loss per share for the periods presented because including them would have been antidilutive for the periods ended:
| September 30, 2019 | September 30, 2018 | |||||||
| (Unaudited) | (Unaudited) | |||||||
| Convertible notes | 9,490,186 | - | ||||||
| Common stock options | 40,438,425 | 39,496,761 | ||||||
| Common stock warrants | 102,758,031 | 118,148,846 | ||||||
| Total common stock equivalents | 152,686,642 | 157,645,607 | ||||||
NOTE 16 – LEASES
Manoa Innovation Center
The Company entered into an automatically renewable month-to-month lease for office space on August 13, 2010. Under the terms of this lease, the Company must provide a written notice 45 days prior to vacating the premises. Total rent expense under this agreement as amended was $8,989 and $27,188 for the three and nine-months ended September 30, 2019, respectively, and $8,760 and $29,662 for the three and nine-months ended September 30, 2018, respectively.
Fleet Lease
In January 2018, the Company entered into a vehicle lease arrangement with a rental company for three vehicles. The terms of the leases require monthly payments of $1,619 for three years. These leases convert to month-to-month leases in January 2021 unless terminated. Total lease expense under this agreement was $4,964 and $16,520 for the three and nine-months ended September 30, 2019, respectively, and $5,602 and $14,953 for the three and nine-months ended September 30, 2018, respectively.
Right-to-use leased asset and liability
As a result of the adoption of ASU No. 2016-02, Leases, on January 1, 2019, the Company recognized a right-to-use leased asset and liability for the Fleet Leases. The balance of this right-to-use asset and liability was $22,015 as of September 30, 2019.
| F-50 |
Cardax, Inc., and Subsidiary
NOTES TO THE CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS (continued)
NOTE 17 – SUBSEQUENT EVENTS
The Company evaluated all material events through the date the financials were ready for issuance and identified the following for additional disclosure.
Convertible Notes
On the dates set forth in the table below , the Company entered into convertible notes with lenders, who are also current stockholders, for the amounts set forth in the table below. Each of these notes accrues interest payable monthly at the rate of 8% per annum and matures on June 30, 2020. Each of these notes and accrued interest thereon may convert into shares of common stock at the conversion price set forth in the table below any time at the holder’s option. If any of these notes, or any portion thereof, has not been repaid or converted in full on or prior to the maturity date, then repayment of the unpaid principal balance plus any accrued and unpaid interest thereon, shall be amortized over the following thirty-six (36) months. The Company has the right to prepay each of these notes without penalty or premium. Each of these notes were issued with detachable five-year warrants to purchase shares of common stock as set forth in the table below.
| Issuance Date | Principal Amount | Original Issue Discount | Gross Proceeds | Note Conversion Price Per Share | Number of Shares Underlying Warrants | Warrant Exercise Price Per Share | ||||||||||||||||||
| October 3, 2019 | $ | 27,174 | $ | 2,174 | $ | 25,000 | $ | 0.12 | 50,000 | $ | 0.12 | |||||||||||||
| October 10, 2019 | 27,174 | 2,174 | 25,000 | 0.12 | 50,000 | 0.12 | ||||||||||||||||||
| October 23, 2019 | 108,696 | 8,696 | 100,000 | 0.12 | 250,000 250,000 |
0.15 0.20 |
||||||||||||||||||
| October 29, 2019 | 27,174 | 2,174 | 25,000 | 0.12 | 50,000 | 0.12 | ||||||||||||||||||
| November 8, 2019 | 16,304 | 1,304 | 15,000 | 0.07 | 30,000 | 0.07 | ||||||||||||||||||
| Total | $ | 206,522 | $ | 16,522 | $ | 190,000 | $ | 0.07-0.12 | 680,000 | $ | 0.07-0.20 | |||||||||||||
On the date set forth in the table below, the Company entered into a senior convertible note payable with a lender, who is also a current stockholder and beneficial owner of more than 5% of the Company’s common stock, in the amount set forth in the table below. This note accrues interest payable monthly at the rate of 8% per annum and matures on June 30, 2020. This note and accrued interest thereon may convert into shares of common stock at the conversion price then in effect (initially $0.12 per share, subject to adjustment) any time at the holder’s option or automatically upon a qualified financing of at least $5 million at the lower of the conversion price then in effect or a 25% discount to the offering price. The conversion price is subject to adjustment upon the issuance of the Company’s common stock or securities convertible into common stock at a price per share less than the then prevailing conversion price, other than specified exempt issuances; accordingly, on November 8, 2019, the conversion price was adjusted to $0.07 per share. The Company has the right to prepay this note without penalty or premium. This note was issued with a detachable five-year warrant to purchase shares of common stock as set forth in the table below. The exercise price of this warrant shall be adjusted in accordance with any adjustment to the conversion price of this note; accordingly, on November 8, 2019, the exercise price was adjusted to $0.07 per share.
| Issuance Date | Principal Amount | Original Issue Discount | Gross Proceeds | Note Conversion Price Per Share | Number of Shares Underlying Warrants | Warrant Exercise Price Per Share | ||||||||||||||||||
| October 16, 2019 | $ | 217,391 | $ | 17,391 | $ | 200,000 | $ | 0.07 | 400,000 | $ | 0.07 | |||||||||||||
General Nutrition Corporation
On October 16, 2019, the exclusivity provision of the Company’s purchasing agreement with GNC expired, however, all other provisions of the Company’s purchasing agreement with GNC remain in effect. The Company may expand ZanthoSyn® distribution to mass market retailers, other specialty nutrition stores, pharmacies, and other retailers. The Company also plans to increase its sales and marketing efforts through e-commerce.
***
| F-51 |
Through and including , 2019, all dealers effecting transactions in the registered securities that may be offered hereby, whether or not participating in this offering, may be required to deliver a prospectus. This delivery requirement is in addition to the obligation of dealers to deliver a prospectus when acting as an underwriter and with respect to an unsold allotment or subscription.
Shares of our common stock
![]()
Sole Book-Running Manager
Maxim Group LLC
PROSPECTUS
, 2019
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution
The following table sets forth the costs and expenses expected to be incurred by Cardax, Inc. (the “Registrant”) in connection with this offering described in this registration statement. All amounts shown are estimates, except the SEC registration fee.
| Item | Amount to be Paid | |||
| SEC registration fee | $ | 4,388.38 | ||
| Legal fees and expenses | 300,000.00 | |||
| Nasdaq entry fee | 50,000.00 | |||
| Accounting fees and expenses | 5,000.00 | |||
| Printing and engraving expenses | 5,000.00 | |||
| Transfer agent fees | 5,000.00 | |||
| Blue sky fees and expenses | 5,000.00 | |||
| Miscellaneous | 10,000.00 | |||
| Total | $ | 384,388.38 | ||
Item 14. Indemnification of Directors and Officers
Our amended and restated certificate of incorporation and bylaws limit our directors’ and officers’ liability to the fullest extent permitted under Delaware corporate law. Specifically, our directors and officers are not liable to us or our stockholders for monetary damages for any breach of fiduciary duty by a director or officer, except for liability:
| ● | for any breach of the director’s or officer’s duty of loyalty to us or our stockholders; | |
| ● | for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law; | |
| ● | under Section 174 of the Delaware General Corporation Law; or | |
| ● | for any transaction from which a director or officer derives an improper personal benefit. |
If the Delaware General Corporation Law is amended to authorize corporate action further eliminating or limiting the personal liability of directors or officers, then the liability of our directors or officers shall be eliminated or limited to the fullest extent permitted by the Delaware General Corporation Law, as so amended.
The provision regarding indemnification of our directors and officers in our amended and restated certificate of incorporation generally does not limit liability under state or federal securities laws.
Delaware law and our amended and restated certificate of incorporation and bylaws provide that we will, in certain situations, indemnify any person made or threatened to be made a party to a proceeding by reason of that person’s former or present official capacity with our Company against judgments, penalties, fines, settlements, and reasonable expenses including reasonable attorney’s fees. Any person is also entitled, subject to certain limitations, to payment or reimbursement of reasonable expenses in advance of the final disposition of the proceeding.
The limitation of liability and indemnification provisions in our amended and restated certificate of incorporation may discourage stockholders from bringing a lawsuit against directors for breach of their fiduciary duty. These provisions may also have the effect of reducing the likelihood of derivative litigation against directors and officers, even though such an action, if successful, might otherwise benefit us and our stockholders. In addition, your investment may be adversely affected to the extent that, in a class action or direct suit, we pay the costs of settlement and damage awards against directors and officers pursuant to these indemnification provisions.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers, or persons controlling the Company pursuant to Delaware law, we are informed that in the opinion of the Securities and Exchange Commission, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
| II-1 |
Item 15. Recent Sales of Unregistered Securities
We issued shares of our common stock or securities convertible into common stock in the following transactions and used the net proceeds for our general working capital and to fund our research, development, and clinical programs.
The securities were issued in reliance upon exemptions from registration pursuant to Section 4(2) of the Securities Act and the rules and regulations promulgated thereunder.
November 2019 Convertible Note
On November 15, 2019, we entered into a convertible note payable with our Chief Executive Officer, as a lender to the Company, in the amount of $100,000. This note accrues interest payable monthly at the rate of 14% per annum and matures on June 30, 2020. This note and accrued interest thereon may convert into shares of our common stock at $0.10 per share any time at the holder’s option. If this note, or any portion thereof, has not been repaid or converted in full on or prior to the maturity date, then repayment of the unpaid principal balance plus any accrued and unpaid interest thereon, shall be amortized over the following thirty-six (36) months. We have the right to prepay this note without penalty or premium.
September-November 2019 Convertible Notes
On the dates set forth in the table below, we entered into convertible notes payable with lenders, who are also current stockholders, in the amounts set forth in the table below. Each of these notes accrues interest payable monthly at the rate of 8% per annum and matures on June 30, 2020. Each of these notes and accrued interest thereon may convert into shares of our common stock at the conversion price set forth in the table below any time at the holder’s option. If any of these notes, or any portion thereof, has not been repaid or converted in full on or prior to the maturity date, then repayment of the unpaid principal balance plus any accrued and unpaid interest thereon, shall be amortized over the following thirty-six (36) months. We have the right to prepay each of these notes without penalty or premium. Each of these notes were issued with detachable five-year warrants to purchase shares of our common stock as set forth in the table below.
| Issuance Date | Principal Amount | Original Issue Discount | Gross Proceeds | Note Conversion Price Per Share | Number of Shares Underlying Warrants |
Warrant Exercise Price Per Share | ||||||||||||||||||
| September 4, 2019 | $ | 108,696 | $ | 8,696 | $ | 100,000 | $ | 0.12 | 200,000 | $ | 0.12 | |||||||||||||
| September 25, 2019 | 54,348 | 4,348 | 50,000 | 0.12 | 100,000 | 0.12 | ||||||||||||||||||
| October 3, 2019 | 27,174 | 2,174 | 25,000 | 0.12 | 50,000 | 0.12 | ||||||||||||||||||
| October 10, 2019 | 27,174 | 2,174 | 25,000 | 0.12 | 50,000 | 0.12 | ||||||||||||||||||
| October 23, 2019 | 108,696 | 8,696 | 100,000 | 0.12 | 250,000 | 0.15 | ||||||||||||||||||
| 250,000 | 0.20 | |||||||||||||||||||||||
| October 29, 2019 | 27,174 | 2,174 | 25,000 | 0.12 | 50,000 | 0.12 | ||||||||||||||||||
| November 8, 2019 | 16,304 | 1,304 | 15,000 | 0.07 | 30,000 | 0.07 | ||||||||||||||||||
| Total | $ | 369,566 | $ | 29,566 | $ | 340,000 | $ | 0.07-0.12 | 980,000 | $ | 0.07-0.20 | |||||||||||||
July-October 2019 Senior Convertible Notes
On the dates set forth in the table below, we entered into senior convertible notes payable with a lender, who is also a current stockholder and beneficial owner of more than 5% of our common stock (prior to the closing of this offering), in the amounts set forth in the table below. Each of these notes accrue interest payable monthly at the rate of 8% per annum and mature on June 30, 2020. Each of these notes and accrued interest thereon may convert into shares of our common stock at the conversion price then in effect (initially $0.12 per share, subject to adjustment) any time at the holder’s option or automatically upon a qualified financing of at least $5 million at the lower of the conversion price then in effect or a 25% discount to the offering price. The conversion price is subject to adjustment upon the issuance of our common stock or securities convertible into our common stock at a price per share less than the then prevailing conversion price, other than specified exempt issuances; accordingly, on November 8, 2019, the conversion price was adjusted to $0.07 per share. We have the right to prepay each of these notes without penalty or premium. Each of these notes were issued with detachable five-year warrants to purchase shares of our common stock as set forth in the table below. The exercise price of these warrants shall be adjusted in accordance with any adjustment to the conversion price of these notes; accordingly, on November 8, 2019, the exercise price was adjusted to $0.07 per share.
| Issuance Date | Principal Amount | Original Issue Discount | Gross Proceeds | Note Conversion Price Per Share | Number of Shares Underlying Warrants | Warrant Exercise Price Per Share | ||||||||||||||||||
| July 19, 2019 | $ | 815,217 | $ | 65,217 | $ | 750,000 | $ | 0.07 | 1,500,000 | $ | 0.07 | |||||||||||||
| October 16, 2019 | 217,391 | 17,391 | 200,000 | 0.07 | 400,000 | 0.07 | ||||||||||||||||||
| Total | $ | 1,032,608 | $ | 82,608 | $ | 950,000 | $ | 0.07 | 1,900,000 | $ | 0.07 | |||||||||||||
April 2019 Convertible Note
On April 18, 2019, we entered into a convertible note payable with a lender in the amount of $150,000. This note accrues interest payable at the rate of 10% per annum and matures on December 31, 2019. This note and accrued interest thereon may convert into shares of our common stock at the conversion price then in effect (initially $0.12 per share, subject to adjustment) any time at the holder’s option or automatically upon maturity provided the 20-day volume weighted average price per share of our common stock upon maturity is at least $0.12 per share. The conversion price is subject to adjustment upon the issuance of our common stock or securities convertible into our common stock at a price per share less than the then prevailing conversion price, other than specified exempt issuances; accordingly, on November 8, 2019, the conversion price was adjusted to $0.07 per share. We have the right to prepay each this note without penalty or premium. This note was issued with a detachable five-year warrant to purchase 500,000 shares of our common stock at $0.20 per share.
2019 Unit Issuances
On February 22, 2019, we sold to a current stockholder of the Company 66,666 units of our securities for an aggregate purchase price of $20,000. Each unit consisted of (i) two shares of our common stock, and (ii) a five-year warrant to purchase one share of our common stock at $0.20. No placement agent or broker dealer was used or participated in any offering or sale of such units.
On March 29, 2019, we sold to a current stockholder of the Company 83,333 units of our securities for an aggregate purchase price of $25,000. Each unit consisted of (i) two shares of our common stock, and (ii) a five-year warrant to purchase one share of our common stock at $0.20. No placement agent or broker dealer was used or participated in any offering or sale of such units.
On April 3, 2019, we sold to a current stockholder of the Company 333,333 units of our securities for an aggregate purchase price of $100,000. Each unit consisted of (i) two shares of our common stock, and (ii) a five-year warrant to purchase one share of our common stock at $0.20. No placement agent or broker dealer was used or participated in any offering or sale of such units.
On April 9, 2019, we sold to a current stockholder of the Company 333,333 units of our securities for an aggregate purchase price of $100,000. Each unit consisted of (i) two shares of our common stock, and (ii) a five-year warrant to purchase one share of our common stock at $0.20. No placement agent or broker dealer was used or participated in any offering or sale of such units.
2017(2) Unit Offering
We sold securities under separate subscription agreements (each, a “2017(2)-Subscription Agreement”), by and between the Company and investors (each a “2017(2)-Purchaser” and collectively, the “2017(2)-Purchasers”), pursuant to which we issued and sold to the 2017(2)-Purchasers units (each a “2017(2)-Unit” and collectively the “2017(2)-Units”) consisting of shares of our common stock and warrants to purchase shares of our common stock.
| II-2 |
During the year ended December 31, 2017, we sold 416,595 2017(2)-Units for an aggregate purchase price of $124,979. Each 2017(2)-Unit consisted of (i) one share of our common stock, and (ii) a five-year warrant to purchase one share of our common stock at $0.30. No placement agent or broker dealer was used or participated in any offering or sale of such 2017(2)-Units.
The foregoing summary of the 2017(2)-Subscription Agreement does not purport to be complete and is qualified in its entirety by reference to the full text of such agreement, which was filed with our Quarterly Report on Form 10-Q filed November 14, 2017.
2017(1) Unit Offering
We sold securities under separate subscription agreements (each, a “2017(1)-Subscription Agreement”), by and between the Company and investors (each a “2017(1)-Purchaser” and collectively, the “2017(1)-Purchasers”), pursuant to which we issued and sold to the 2017(1)-Purchasers units (each a “2017(1)-Unit” and collectively the “2017(1)-Units”) consisting of shares of our common stock and warrants to purchase shares of our common stock.
During the year ended December 31, 2017, we sold an aggregate of 31,453,788 2017(1)-Units for an aggregate purchase price of $3,774,456. Each 2017(1)-Unit consisted of (i) one share of our common stock, and (ii) a five-year warrant to purchase one share of our common stock at $0.12. No placement agent or broker dealer was used or participated in any offering or sale of such 2017(1)-Units.
The foregoing summary of the 2017(1)-Subscription Agreement does not purport to be complete and is qualified in its entirety by reference to the full text of such agreement, which was filed with our Annual Report on Form 10-K filed March 31, 2017.
2016/2017 Unit Offering
We sold securities under separate subscription agreements (each, a “2016/2017-Subscription Agreement”), by and between the Company and investors (each a “2016/2017-Purchaser” and collectively, the “2016/2017-Purchasers”), pursuant to which we issued and sold to the 2016/2017-Purchasers units (each a “2016/2017-Unit” and collectively the “2016/2017-Units”) consisting of shares of our common stock and warrants to purchase shares of our common stock.
During the year ended December 31, 2016 and the first quarter of 2017, we sold an aggregate of 16,250,000 2016/2017-Units for an aggregate purchase price of $1,300,000. Each 2016/2017-Unit consisted of (i) one share of our common stock, (ii) a five-year warrant to purchase one share of our common stock at $0.08, (iii) a five-year warrant to purchase one share of our common stock at $0.12, and (iv) a five-year warrant to purchase one share of our common stock at $0.16. No placement agent or broker dealer was used or participated in any offering or sale of such 2016/2017-Units.
The foregoing summary of the 2016/2017-Subscription Agreement does not purport to be complete and is qualified in its entirety by reference to the full text of such agreement, which was filed with our Quarterly Report on Form 10-Q on May 13, 2016.
Equity Purchase Agreement
On March 7, 2017, we sold 567,644 shares of our common stock at a price of $0.1057 per share pursuant to the equity purchase agreement (the “Equity Purchase Agreement”) with Southridge Partners II LP (“Southridge”), which we previously reported in the Registration Statement on Form S-1 (333-214049) filed on February 8, 2017. Pursuant to the terms of the Equity Purchase Agreement, we have the right, but not the obligation, to sell shares of our common stock to Southridge and Southridge has the right to resell the shares of our common stock.
The foregoing summary of the Equity Purchase Agreement does not purport to be complete and is qualified in its entirety by reference to the full text of such agreement, which was filed with our Current Report on Form 8-K on July 13, 2016.
Settlement of Payable
On May 3, 2017, we settled a payable in the amount of $44,700 with a previously engaged broker dealer through the issuance 558,750 units. Each unit consisted of: (i) one (1) share of our common stock, (ii) a five-year warrant to purchase one (1) share of our common stock at $0.08 per share, (iii) a five-year warrant to purchase one (1) share of our common stock at $0.12 per share, and (iv) a five-year warrant to purchase one (1) share of our common stock at $0.16 per share.
Warrant Exercise
During the year ended December 31, 2017, we issued 233,217 shares of our common stock in connection with the cashless exercise of a warrant for 298,000 shares of our common stock at $0.10 per share with 64,783 shares of our common stock withheld with an aggregate fair market value equal to the aggregate exercise price
During the year ended December 31, 2017, we issued 500,000 shares of our common stock in connection with the exercise of a warrant for 500,000 shares of our common stock at $0.08 per share in exchange for $40,000.
Stock Option Exercise
On April 16, 2018, we issued 76,924 shares of our common stock in connection with the cashless exercise of stock options for 100,000 shares of our common stock at $0.06 per share with 23,076 shares of our common stock withheld with an aggregate fair market value equal to the aggregate exercise price.
On May 2, 2018, we issued 80,073 shares of our common stock in connection with the cashless exercise of stock options for 100,000 shares of our common stock at $0.06 per share with 19,927 shares of our common stock withheld with an aggregate fair market value equal to the aggregate exercise price.
During the year ended December 31, 2017, we issued 645,288 shares of our common stock in connection with the cashless exercise of stock options for 100,000, 45,000, and 625,000 shares of our common stock at $0.155, $0.06, and $0.06, respectively, per share with 124,712 shares of our common stock withheld with an aggregate fair market value equal to the aggregate exercise price.
| II-3 |
Stock Based Compensation
During the year ended December 31, 2017, we issued 793,025 shares of our common stock to our independent directors for compensation and 200,000 shares of our common stock to service providers for compensation. During the year ended December 31, 2018, we issued 1,344,274 shares of our common stock to our independent directors for compensation and 112,500 shares of our common stock to service providers for compensation. From January 1, 2019 to the date of this prospectus, we issued 1,043,858 shares of our common stock to our independent directors for compensation and 75,000 shares of our common stock to service providers for compensation.
On July 27, 2018, we issued warrants to purchase 315,010 shares of our common stock at an exercise price of $0.21 per share until July 27, 2023 to placement agents for compensation.
The following table sets forth options to purchase shares of our common stock we issued to employees, directors, and service providers for compensation.
| Issuance Date | Number of Shares Underlying Options | Option Exercise Price Per Share | Option Expiration Date | Option Vesting | ||||||||||
| March 31, 2017 | 78,125 | $ | 0.185 | March 31, 2022 | (1) | |||||||||
| June 30, 2017 | 83,333 | $ | 0.20 | June 30, 2022 | (1) | |||||||||
| September 25, 2017 | 400,000 | $ | 0.50 | September 25, 2027 | (2) | |||||||||
| September 25, 2017 | 400,000 | $ | 0.47 | September 25, 2027 | (3) | |||||||||
| November 1, 2017 | 1,000,000 | $ | 0.44 | November 1, 2027 | (2) | |||||||||
| November 27, 2017 | 100,000 | $ | 0.37 | November 27, 2027 | (4) | |||||||||
| December 13, 2017 | 100,000 | $ | 0.34 | December 13, 2027 | (2) | |||||||||
| January 1, 2018 | 500,000 | $ | 0.16 | January 1, 2028 | (2) | |||||||||
| January 1, 2018 | 333,334 | $ | 0.16 | January 1, 2023 | (5) | |||||||||
| June 1, 2018 | 1,000,000 | $ | 0.24 | June 1, 2028 | (6) | |||||||||
| November 14, 2018 | 1,000,000 | $ | 0.21 | November 14, 2028 | (7) | |||||||||
| (1) | The shares were fully vested upon issuance. | |
| (2) | One-fourth of the shares vest one year from issuance and one forty-eighth of the shares vest monthly thereafter. | |
| (3) | On January 31, 2018, 50,000 shares were fully vested and 350,000 shares were cancelled. | |
| (4) | On August 16, 2019, 41,664 shares were fully vested and 58,336 shares were cancelled. | |
| (5) | The shares vest monthly over one year. | |
| (6) | The shares vest monthly over two years. | |
| (7) | One-half of the shares vest monthly over four years and the remaining shares vest upon certain milestones. |
| II-4 |
Item 16. Exhibits and Financial Statement Schedules.
(a) Exhibits
| II-5 |
| 10.13 | Form of Class E Warrant(6) | |
| 10.14 | Independent Directors’ Compensation Agreement(7) | |
| 10.15 | Supplement to Senior Executive Employment Agreement of David G. Watumull(7) | |
| 10.16 | Payment Deferral and Acceptance Agreement of JBR Business Solutions, LLC(7) | |
| 10.17 | Form of Payment Deferral and Acceptance Agreement(7) | |
| 10.18 | Form of the Senior Convertible Notes issued by Cardax, Inc., dated July 19, 2019 and October 16, 2019(8) | |
| 10.19 | Form of the Securities Purchase Agreements of Cardax, Inc., dated July 19, 2019 and October 16, 2019(8) | |
| 10.20 | Form of the Warrants issued by Cardax, Inc., dated July 19, 2019 and October 16, 2019(8) | |
| 10.21 | Exclusivity Agreement, dated October 16, 2017, by and between Cardax, Inc. and General Nutrition Corporation(9) | |
| 10.22 | ||
| 10.23 | ||
| 21.1 | Subsidiaries of Cardax, Inc.(3) | |
| 23.1 | Consent of KBL, LLP* | |
| 23.2 | Consent of Allegaert Berger & Vogel LLP (contained in the Opinion of Allegaert Berger & Vogel LLP under Exhibit 5.1) | |
| 24.1 | Power of Attorney# |
| # | Previously filed. |
| * | Filed herein. |
| ** | To be filed by an amendment to this Registration Statement |
| (1) | Filed as an exhibit to the Current Report on Form 8-K of the Company dated November 27, 2013. |
| (2) | Filed as an exhibit to the Current Report on Form 8-K of the Company dated January 13, 2014. |
| (3) | Filed as an exhibit to the Current Report on Form 8-K of the Company dated February 10, 2014. |
| (4) | Filed as an exhibit to the Current Report on Form 8-K of the Company dated November 24, 2015. |
| (5) | Filed as an exhibit to Amendment No. 1 of the Registration Statement on Form S-1 of the Company dated September 2, 2014. |
| (6) | Filed as an exhibit to the Current Report on Form 8-K of the Company filed March 9, 2015. |
| (7) | Filed as an exhibit to the Current Report on Form 8-K of the Company filed July 7, 2015. |
| (8) | Filed as an exhibit to the Quarterly Report on Form 10-Q of the Company filed July 25, 2019. |
| (9) | Filed as an exhibit to the Current Report on Form 8-K of the Company filed November 14, 2017. |
| 101.INS | XBRL Instance Document* | |
| 101.SCH | XBRL Taxonomy Extension Schema Document* | |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document* | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document* | |
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document* | |
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document* |
| II-6 |
(b) Financial Statement Schedules
All financial statement schedules are included in the Registrant’s consolidated financial statements and the related notes thereto, or are inapplicable or otherwise not required.
Item 17. Undertakings
The undersigned Registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) to include any prospectus required by Section 10(a)(3) of the Securities Act of 1933, as amended (the “Act”);
(ii) to reflect in the prospectus any facts or events arising after the effective date of this registration statement (or the most-recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement;
(iii) to include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement.
(2) That, for the purpose of determining any liability under the Act, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) Insofar as indemnification for liabilities arising under the Act may be permitted to directors, officers, and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer, or controlling person of the registrant in the successful defense of any action, suit, or proceeding) is asserted by such director, officer, or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
(5) That, for the purpose of determining liability under the Act, to any purchaser, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
| II-7 |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the Registrant has duly caused this registration statement to be filed on its behalf by the undersigned, thereunto duly authorized in the City and County of Honolulu, State of Hawaii on November 22, 2019.
| CARDAX, INC. | ||
| By: | /s/ David G. Watumull | |
| Name: | David G. Watumull | |
| Title: | President & Chief Executive Officer | |
Pursuant to the requirements of the Securities Act of 1933, this Registration Statement on Form S-1 has been signed by the following persons in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ David G. Watumull | President, Chief Executive Officer, and Director | November 22, 2019 | ||
| David G. Watumull | (president, principal executive officer, and director) | |||
| /s/ John B. Russell | Chief Financial Officer and Treasurer | November 22, 2019 | ||
| John B. Russell | (principal financial officer and treasurer) | |||
| * | Chairman of the Board of Directors | November 22, 2019 | ||
| George W. Bickerstaff, III | ||||
| * | Director | November 22, 2019 | ||
| Terence A. Kelly, Ph.D. | ||||
| * | Director | November 22, 2019 | ||
| Michele Galen | ||||
| * | Director | November 22, 2019 | ||
| Makarand Jawadekar, Ph.D. | ||||
| * | Director | November 22, 2019 | ||
| Elona Kogan |
| * By: | /s/ David G. Watumull | |
| David G. Watumull | ||
| Attorney-in-Fact |
| II-8 |